Cheminformatics
Molecular similarity evaluation methods using spherical harmonics
-
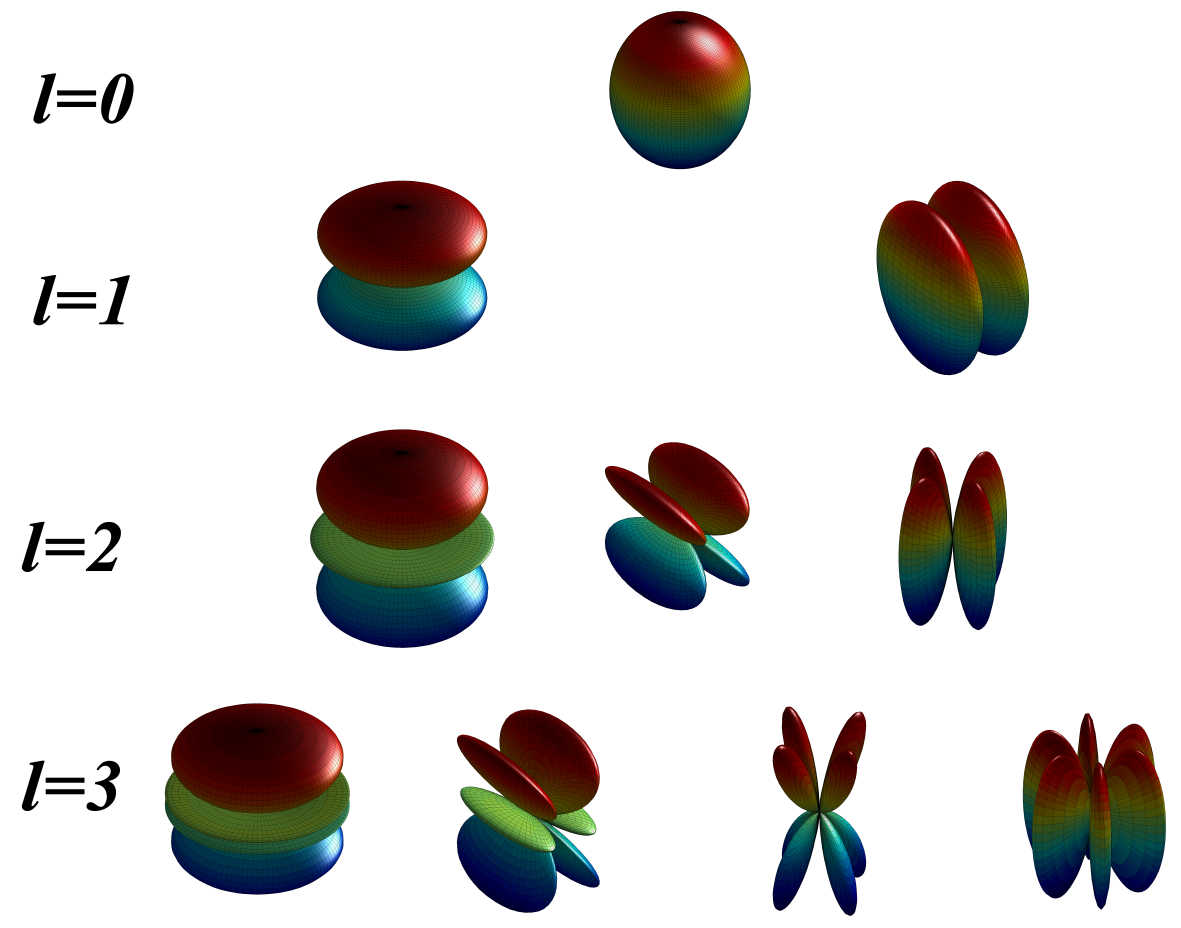 A novel molecular shape similarity comparisonmethod, namely SHeMS, derived from spherical harmonic(SH) expansion, is presented in this study. Through weightoptimization using genetic algorithms for a customizedreference set, the optimal combination of weights for thetranslationally and rotationally invariant (TRI) SH shapedescriptor, which can specifically and effectively distinguish overall and detailed shape features according to themolecular surface, is obtained for each molecule.
A novel molecular shape similarity comparisonmethod, namely SHeMS, derived from spherical harmonic(SH) expansion, is presented in this study. Through weightoptimization using genetic algorithms for a customizedreference set, the optimal combination of weights for thetranslationally and rotationally invariant (TRI) SH shapedescriptor, which can specifically and effectively distinguish overall and detailed shape features according to themolecular surface, is obtained for each molecule. The Complex Structures of hsDHODH and pfDHODH with inhibitors
-
 Pyrimidines and their derivatives, which play important roles in glycogen metabolism, glycerophospholipid metabolism, DNA and RNA biosynthesis, are considered to be essential for cellular metabolism and cell growth. Dihydroorotate dehydrogenase (DHODH) is a key enzyme of de novo pyrimidine synthesis. Due to its significant biological functions, DHODH is considered to be a safe and effective target for the treatment of cancer, autoimmune diseases, malaria and so on. Therefore, the discovery of novel DHODH inhibitors has become a hot research topic at present.
Pyrimidines and their derivatives, which play important roles in glycogen metabolism, glycerophospholipid metabolism, DNA and RNA biosynthesis, are considered to be essential for cellular metabolism and cell growth. Dihydroorotate dehydrogenase (DHODH) is a key enzyme of de novo pyrimidine synthesis. Due to its significant biological functions, DHODH is considered to be a safe and effective target for the treatment of cancer, autoimmune diseases, malaria and so on. Therefore, the discovery of novel DHODH inhibitors has become a hot research topic at present. SimG: An Alignment Based Method for Evaluating the Similarity of Small Molecules and Binding Sites
-
 In this study, a Gaussian volume overlap and chemical feature based molecular similarity metric was devised, and a downhill simplex searching was carried out to evaluate the corresponding similarity. By representing the shapes of both the candidate small molecules and the binding site with chemical features and comparing the corresponding Gaussian volumes overlaps, the active compounds could be identified. These two aspects compose the proposed method named SimG which supports both structure-based and ligand-based strategies. The validity of the proposed method was examined by analyzing the similarity score variation between actives and decoys as well as correlation among distinct reference methods. A retrospective virtual screening test was carried out on DUD datasets, demonstrating that the performance of structure-based shape matching virtual screening in DUD datasets is substantially dependent on some physical properties, especially the solvent-exposure extent of the binding site: The enrichments of targets with less solvent-exposed binding sites generally exceeds that of the one with more solvent-exposed binding sites and even surpasses the corresponding ligand-based virtual screening. J. Chem. Inf. Model., 2013, 53 (8), pp 2103–2115 DOI: 10.1021/ci400139j
In this study, a Gaussian volume overlap and chemical feature based molecular similarity metric was devised, and a downhill simplex searching was carried out to evaluate the corresponding similarity. By representing the shapes of both the candidate small molecules and the binding site with chemical features and comparing the corresponding Gaussian volumes overlaps, the active compounds could be identified. These two aspects compose the proposed method named SimG which supports both structure-based and ligand-based strategies. The validity of the proposed method was examined by analyzing the similarity score variation between actives and decoys as well as correlation among distinct reference methods. A retrospective virtual screening test was carried out on DUD datasets, demonstrating that the performance of structure-based shape matching virtual screening in DUD datasets is substantially dependent on some physical properties, especially the solvent-exposure extent of the binding site: The enrichments of targets with less solvent-exposed binding sites generally exceeds that of the one with more solvent-exposed binding sites and even surpasses the corresponding ligand-based virtual screening. J. Chem. Inf. Model., 2013, 53 (8), pp 2103–2115 DOI: 10.1021/ci400139j
Target Identification
Enhancing the Enrichment of Pharmacophore-Based Target Prediction
-
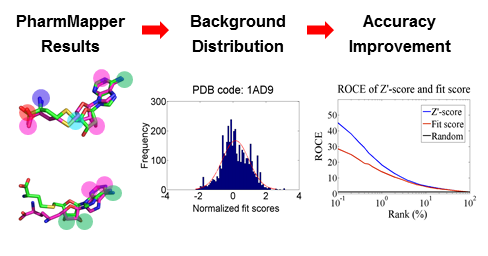 We improved the predictive accuracy of PharmMapper by generating a ligand–target pairwise fit score matrix from profiling all the annotated pharmacophore models against corresponding ligands in the original complex structures that were used to extract these pharmacophore models. The matrix reflects the noise baseline of fit score distribution of the background database, thus enabling estimation of the probability of finding a given target randomly with the calculated ligand–pharmacophore fit score.
We improved the predictive accuracy of PharmMapper by generating a ligand–target pairwise fit score matrix from profiling all the annotated pharmacophore models against corresponding ligands in the original complex structures that were used to extract these pharmacophore models. The matrix reflects the noise baseline of fit score distribution of the background database, thus enabling estimation of the probability of finding a given target randomly with the calculated ligand–pharmacophore fit score.
Network Pharmacology
Predicting the Drug Safety for Traditional Chinese Medicine
-
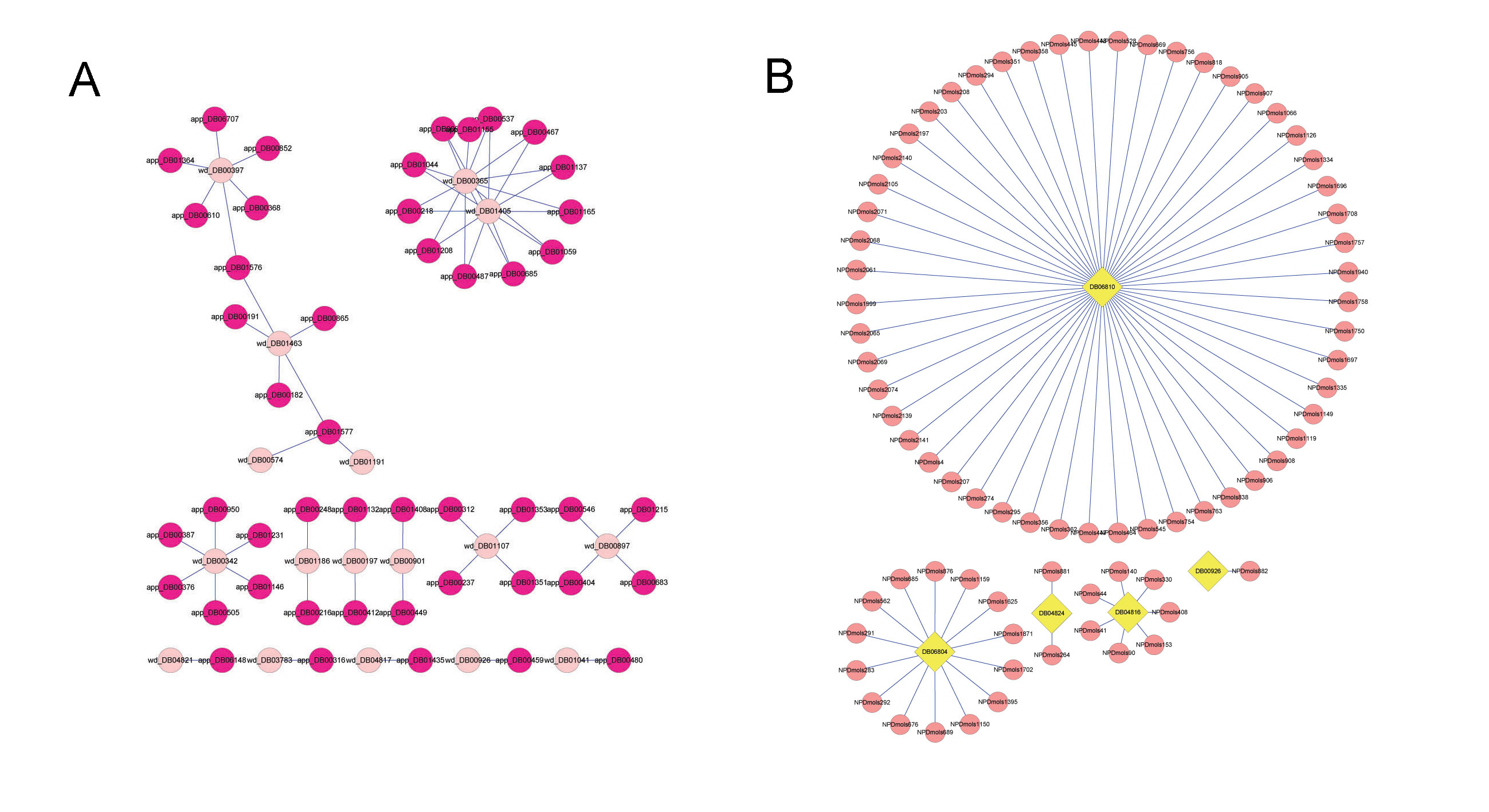 Here, we integrated a network topology analysis with cheminformatics measurements on drug information from the DrugBank database to detect the discrepancies between approved drugs and withdrawn drugs and give drug safety indications.
Here, we integrated a network topology analysis with cheminformatics measurements on drug information from the DrugBank database to detect the discrepancies between approved drugs and withdrawn drugs and give drug safety indications.
Drug Discovery
Rational Design of Potent Inhibitors of hDHODH with in Vivo Anti-arthritic Activity
-
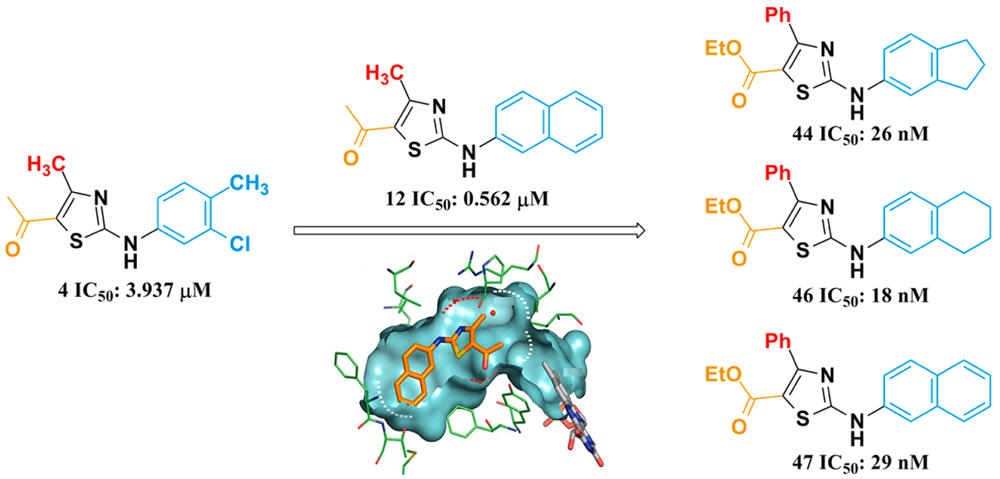
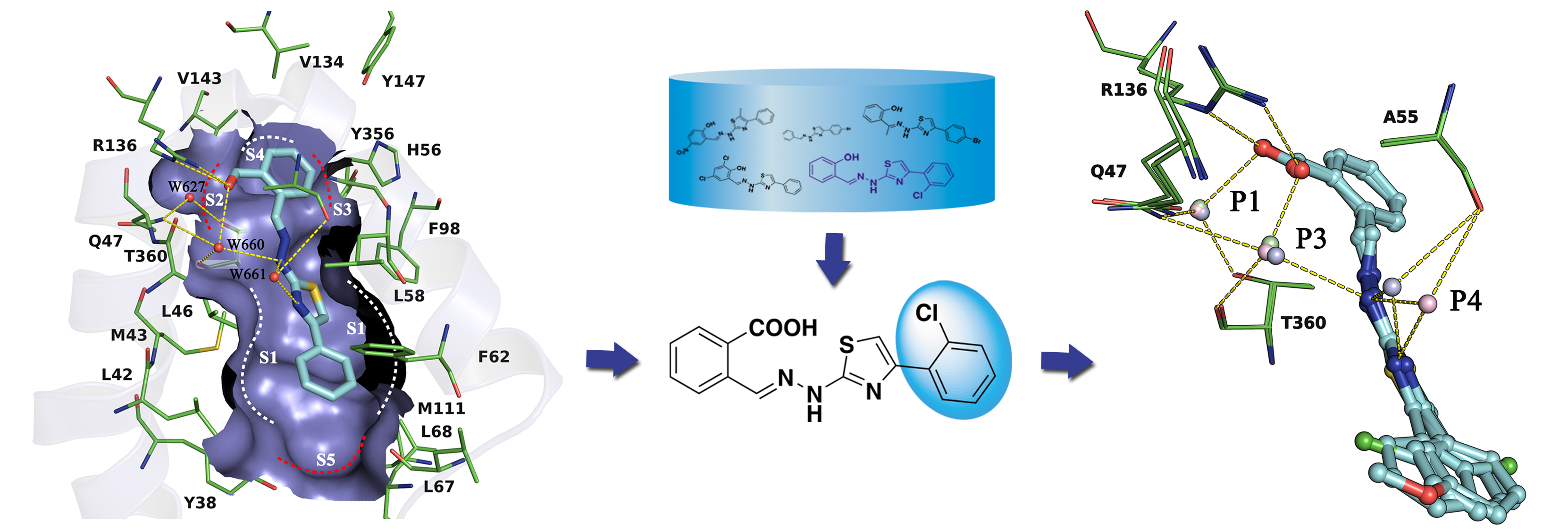 Human dihydroorotate dehydrogenase (hDHODH) is an attractive therapeutic target for the treatment of rheumatoid arthritis, transplant rejection and other autoimmune diseases. Based on the X-ray structure of hDHODH in complex with lead compound 7, a series of benzylidenehydrazinyl-substituted thiazole derivatives as potent inhibitors of hDHODH were designed and synthesized, of which 19 and 30 were the most potent with IC50 values in the double-digit nanomolar range. Moreover, compound 19 displayed significant anti-arthritic effects and favorable pharmacokinetic profiles in vivo. Further X-ray structure and SAR analyses revealed that the potencies of the designed inhibitors were partly attributable to additional water-mediated hydrogen bond networks formed by an unexpected buried water between hDHODH and the 2-(2-methylenehydrazinyl)thiazole scaffold. This work not only elucidates promising scaffolds targeting hDHODH for the treatment of rheumatoid arthritis, but also demonstrates that the water-mediated hydrogen bond interaction is an important factor in molecular design and optimization.
Human dihydroorotate dehydrogenase (hDHODH) is an attractive therapeutic target for the treatment of rheumatoid arthritis, transplant rejection and other autoimmune diseases. Based on the X-ray structure of hDHODH in complex with lead compound 7, a series of benzylidenehydrazinyl-substituted thiazole derivatives as potent inhibitors of hDHODH were designed and synthesized, of which 19 and 30 were the most potent with IC50 values in the double-digit nanomolar range. Moreover, compound 19 displayed significant anti-arthritic effects and favorable pharmacokinetic profiles in vivo. Further X-ray structure and SAR analyses revealed that the potencies of the designed inhibitors were partly attributable to additional water-mediated hydrogen bond networks formed by an unexpected buried water between hDHODH and the 2-(2-methylenehydrazinyl)thiazole scaffold. This work not only elucidates promising scaffolds targeting hDHODH for the treatment of rheumatoid arthritis, but also demonstrates that the water-mediated hydrogen bond interaction is an important factor in molecular design and optimization. Novel Selective Inhibitors of PfDHODH: Discovery and Optimization of Dihydrothiophenone Derivatives
-
 Taking the emergence of drug resistance and lack of effective antimalarial vaccines into consideration, it is of significant importance to develop novel antimalarial agents for the treatment of malaria. Herein, we elucidated the discovery and structure–activity relationships of a series of dihydrothiophenone derivatives as novel specific inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase (PfDHODH). The most promising compound, 50, selectively inhibited PfDHODH (IC50 = 6 nM, with >14 000-fold species-selectivity over hDHODH) and parasite growth in vitro (IC50 = 15 and 18 nM against 3D7 and Dd2 cells, respectively). Moreover, an oral bioavailability of 40% for compound 50 was determined from in vivo pharmacokinetic studies. These results further indicate that PfDHODH is an effective target for antimalarial chemotherapy, and the novel scaffolds reported in this work might lead to the discovery of new antimalarial agents.
Taking the emergence of drug resistance and lack of effective antimalarial vaccines into consideration, it is of significant importance to develop novel antimalarial agents for the treatment of malaria. Herein, we elucidated the discovery and structure–activity relationships of a series of dihydrothiophenone derivatives as novel specific inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase (PfDHODH). The most promising compound, 50, selectively inhibited PfDHODH (IC50 = 6 nM, with >14 000-fold species-selectivity over hDHODH) and parasite growth in vitro (IC50 = 15 and 18 nM against 3D7 and Dd2 cells, respectively). Moreover, an oral bioavailability of 40% for compound 50 was determined from in vivo pharmacokinetic studies. These results further indicate that PfDHODH is an effective target for antimalarial chemotherapy, and the novel scaffolds reported in this work might lead to the discovery of new antimalarial agents. Biological evaluation of quinoline derivatives as inhibitors of human dihydroorotate dehydrogenase
-
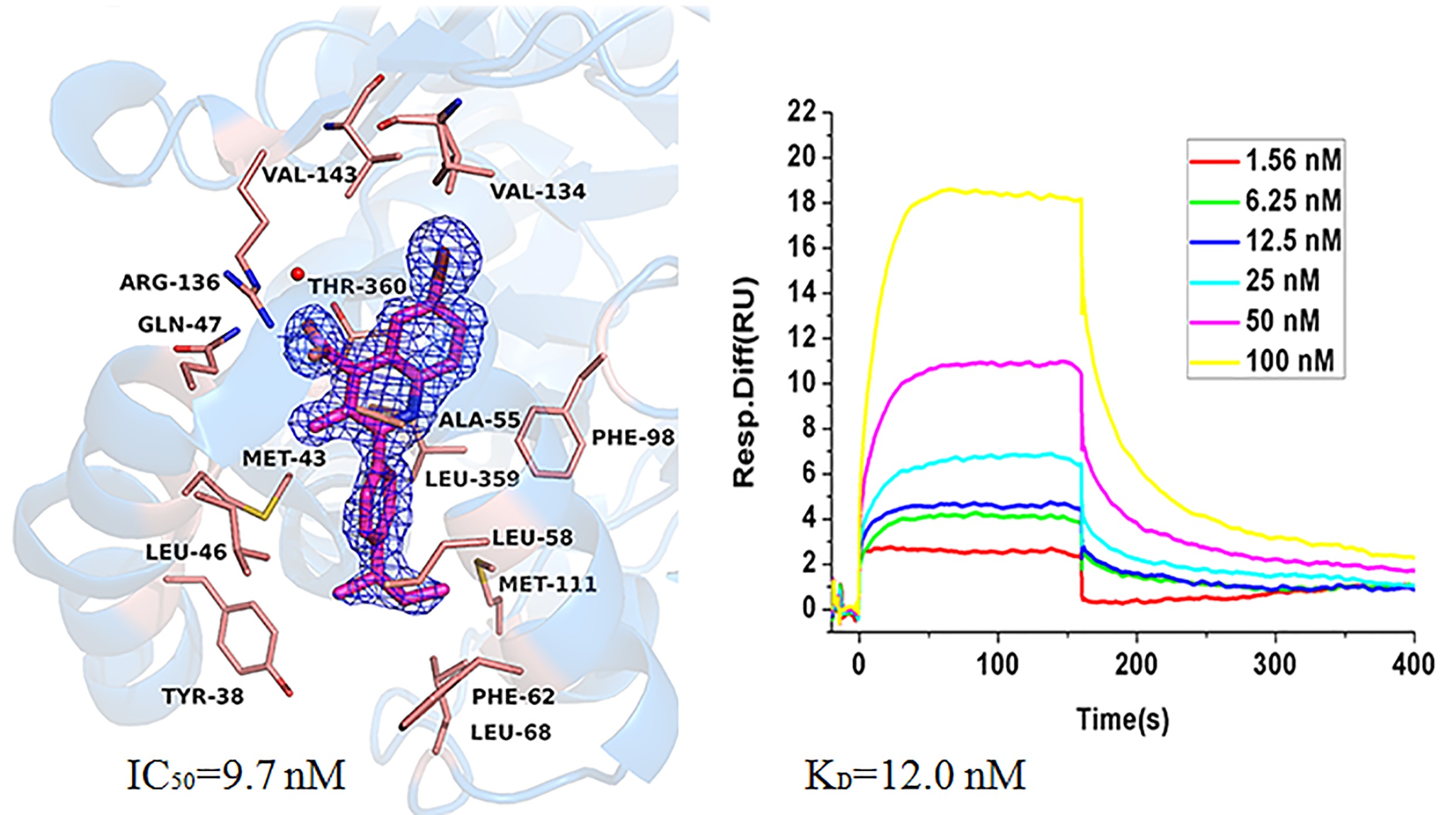 Human dihydroorotate dehydrogenase (hDHODH) is an enzyme that catalyzes the fourth step in de novo pyrimidine biosynthesis, and its inhibitors restrict the growth of rapidly proliferating cells. Therefore, hDHODH has been reported as an attractive target for the treatment of cancer and autoimmune diseases. In this study, several quinoline derivatives were identified as potent inhibitors against hDHODH, among which compound A9 was the most potent one with an IC50 value of 9.7 nM. We further verified that A9 could directly bind to the target hDHODH by thermal shift assay (TSA), surface plasmon resonance (SPR) and X-ray crystallography. Moreover, the binding mode of compound A9 and structure-activity relationship (SAR) of quinoline derivatives with hDHODH were summarized.
Human dihydroorotate dehydrogenase (hDHODH) is an enzyme that catalyzes the fourth step in de novo pyrimidine biosynthesis, and its inhibitors restrict the growth of rapidly proliferating cells. Therefore, hDHODH has been reported as an attractive target for the treatment of cancer and autoimmune diseases. In this study, several quinoline derivatives were identified as potent inhibitors against hDHODH, among which compound A9 was the most potent one with an IC50 value of 9.7 nM. We further verified that A9 could directly bind to the target hDHODH by thermal shift assay (TSA), surface plasmon resonance (SPR) and X-ray crystallography. Moreover, the binding mode of compound A9 and structure-activity relationship (SAR) of quinoline derivatives with hDHODH were summarized. 4-thiazolidinone derivatives as novel inhibitors of hDHODH
-
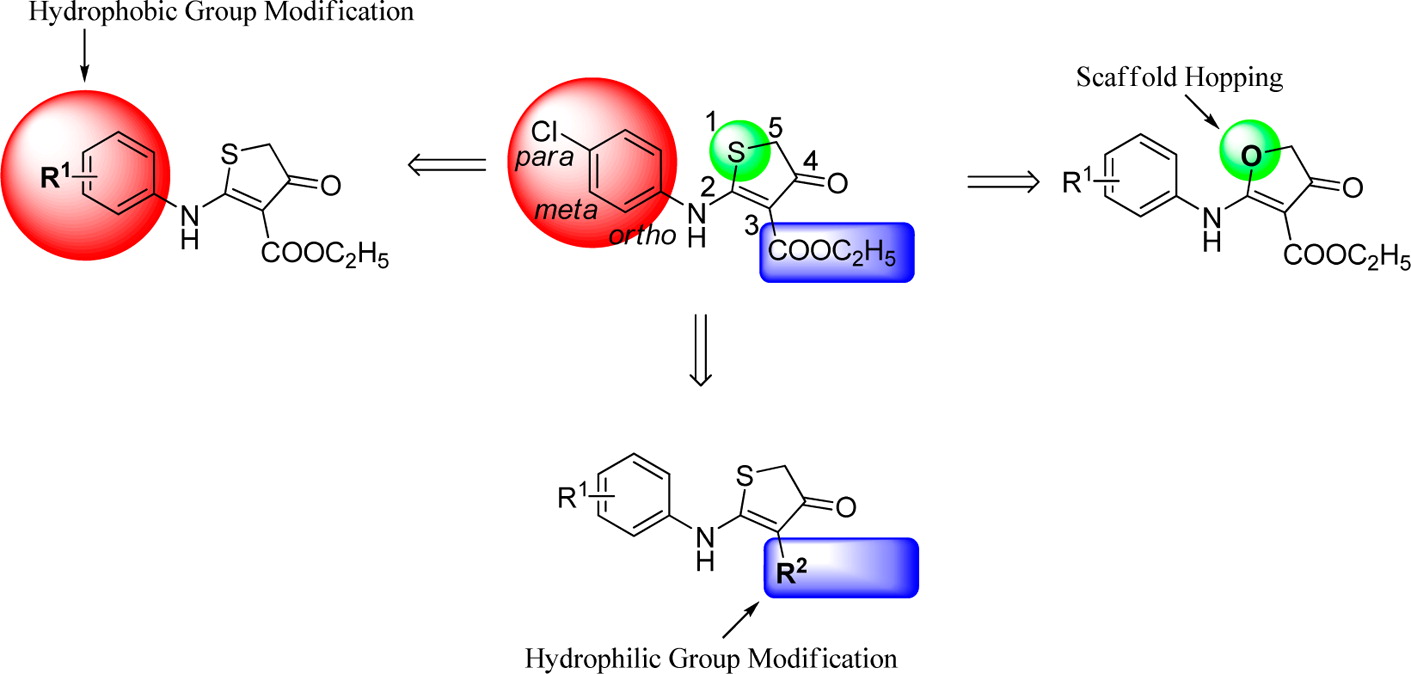
A series of 4-thiazolidinone derivatives were synthesized and evaluated as novel human dihydroorotate dehydrogenase (hDHODH) inhibitors. Compounds 26 and 31 displayed IC50 values of 1.75 and 1.12 μM, respectively. The structure–activity relationship was summarized. Further binding mode analysis revealed that compound 31 could form a hydrogen bond with Tyr38 and a water-mediated hydrogen bond with Ala55, which may be necessary for maintaining the bioactivities of the compounds in this series. Further structural optimization of the para- or meta-position of the phenyl group at R will lead to the identification of more potent hDHODH inhibitors.
Discovery and Rational Design of Pteridin-7(8H)-one-based Inhibitors Targeting FLT3 and Its Mutants
-
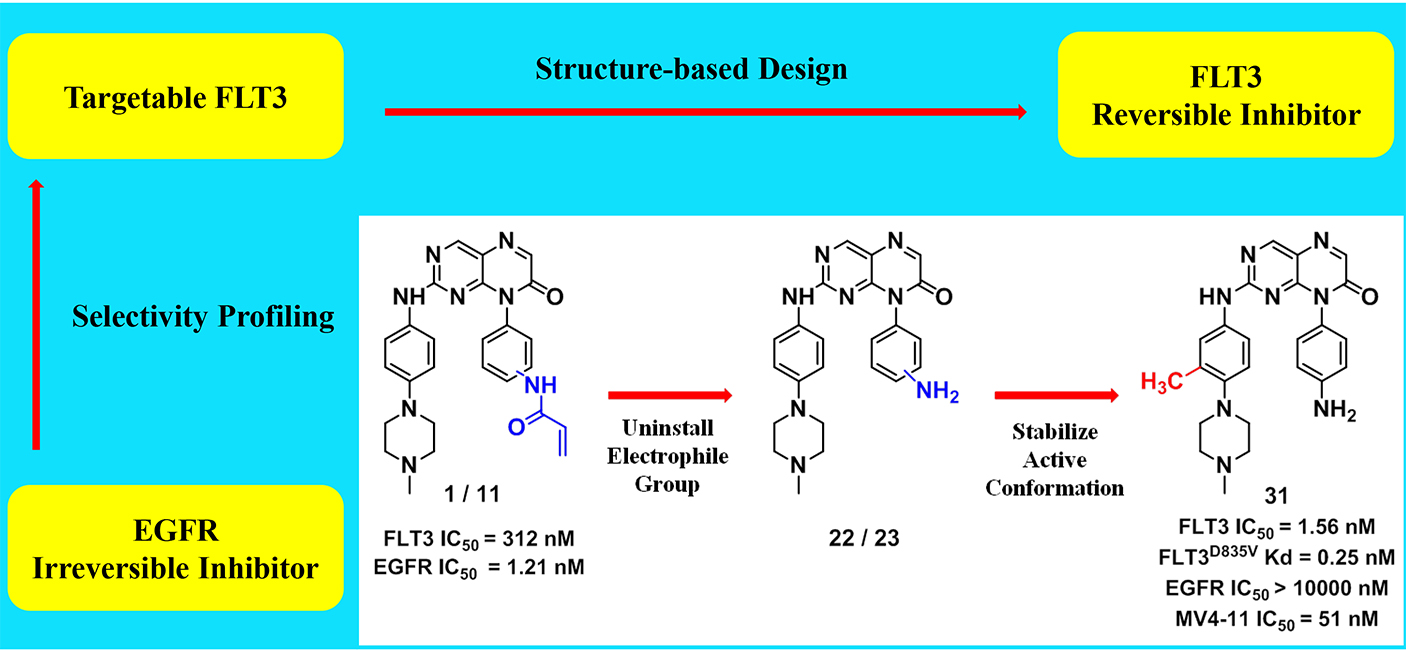 FLT3 has been validated as a therapeutic target for the treatment of acute myeloid leukemia (AML). In this paper, we describe for the first time, pteridin-7(8H)-one as a scaffold for potent FLT3 inhibitors derived from structural optimizations on irreversible EGFR inhibitors. The representative inhibitor (31) demonstrates single-digit nanomolar inhibition against FLT3, subnanomolar KD for drug-resistance FLT3 mutants. In profiling of the in vitro tumor cell lines, it shows good selectivity against AML cells harboring FLT3-ITD mutations over other leukemia and solid tumor cell lines. The mechanism of action study illustrates that pteridin-7(8H)-one derivatives suppress the phosphorylation of FLT3 and its downstream pathways, thereby inducing G0/G1 cell cycle arrest and apoptosis in AML cells. In in vivo studies, 31 significantly suppresses the tumor growth in MV4-11 xenograft model. Overall, we provide a structurally distinct chemical scaffold with which to develop FLT3-mutants-selective inhibitors for AML treatment.
FLT3 has been validated as a therapeutic target for the treatment of acute myeloid leukemia (AML). In this paper, we describe for the first time, pteridin-7(8H)-one as a scaffold for potent FLT3 inhibitors derived from structural optimizations on irreversible EGFR inhibitors. The representative inhibitor (31) demonstrates single-digit nanomolar inhibition against FLT3, subnanomolar KD for drug-resistance FLT3 mutants. In profiling of the in vitro tumor cell lines, it shows good selectivity against AML cells harboring FLT3-ITD mutations over other leukemia and solid tumor cell lines. The mechanism of action study illustrates that pteridin-7(8H)-one derivatives suppress the phosphorylation of FLT3 and its downstream pathways, thereby inducing G0/G1 cell cycle arrest and apoptosis in AML cells. In in vivo studies, 31 significantly suppresses the tumor growth in MV4-11 xenograft model. Overall, we provide a structurally distinct chemical scaffold with which to develop FLT3-mutants-selective inhibitors for AML treatment. Identification of RSK2 inhibitors
-
 We focused on the RSK2 kinase to do corresponding drug development including discovery of RSK2 inhibitors by a ligand-based virtual screening, structural modifications, structure-activity relationship explorations and potential binding modes investigations on RSK2 inhibitors, which contributed to abundant RSK2 inhibitors with both potent activities and novel structures, especially some hits showed superior selectivity against RSK2 and moderate anti-proliferation effects on human breast cancer cells and prostate cancer cells.
We focused on the RSK2 kinase to do corresponding drug development including discovery of RSK2 inhibitors by a ligand-based virtual screening, structural modifications, structure-activity relationship explorations and potential binding modes investigations on RSK2 inhibitors, which contributed to abundant RSK2 inhibitors with both potent activities and novel structures, especially some hits showed superior selectivity against RSK2 and moderate anti-proliferation effects on human breast cancer cells and prostate cancer cells. Discovery of Inhibitors targeting EGFR Kinase T790M/L858R mutant
-
 The EGFR T790M variant is an important mutation, resulting in approximately 50% of the clinically acquired resistance to approved EGFR inhibitors. Starting with a previously reported pyrimidine-based EGFR inhibitor, a novel pteridin-7(8H)-one scaffold with a high 3D similarity was found and transformed into irreversible inhibitors of the EGFR T790M mutant. The most potent compounds, 3q and 3x, exhibited excellent enzyme inhibitory activities, with subnanomolar IC50 values for both the wild-type and T790M/L858R double mutant EGFRs, as well as potent cellular antiproliferative activities against both gefitinib-sensitive and -resistant cancer cell lines. The in vivo antitumor efficacy study demonstrated that compound 3x significantly inhibited tumor growth and induced tumor stasis in an EGFR-T790M/L858R-driven human nonsmall-cell lung cancer xenograft mouse model. This work demonstrated the utility of this sophisticated computational design strategy for fast 3D scaffold hopping with competitive bioactivities to meet an important clinical need.
The EGFR T790M variant is an important mutation, resulting in approximately 50% of the clinically acquired resistance to approved EGFR inhibitors. Starting with a previously reported pyrimidine-based EGFR inhibitor, a novel pteridin-7(8H)-one scaffold with a high 3D similarity was found and transformed into irreversible inhibitors of the EGFR T790M mutant. The most potent compounds, 3q and 3x, exhibited excellent enzyme inhibitory activities, with subnanomolar IC50 values for both the wild-type and T790M/L858R double mutant EGFRs, as well as potent cellular antiproliferative activities against both gefitinib-sensitive and -resistant cancer cell lines. The in vivo antitumor efficacy study demonstrated that compound 3x significantly inhibited tumor growth and induced tumor stasis in an EGFR-T790M/L858R-driven human nonsmall-cell lung cancer xenograft mouse model. This work demonstrated the utility of this sophisticated computational design strategy for fast 3D scaffold hopping with competitive bioactivities to meet an important clinical need. Discovery and optimization of 2-aminopyridine derivatives as novel and selective JAK2 inhibitors
-
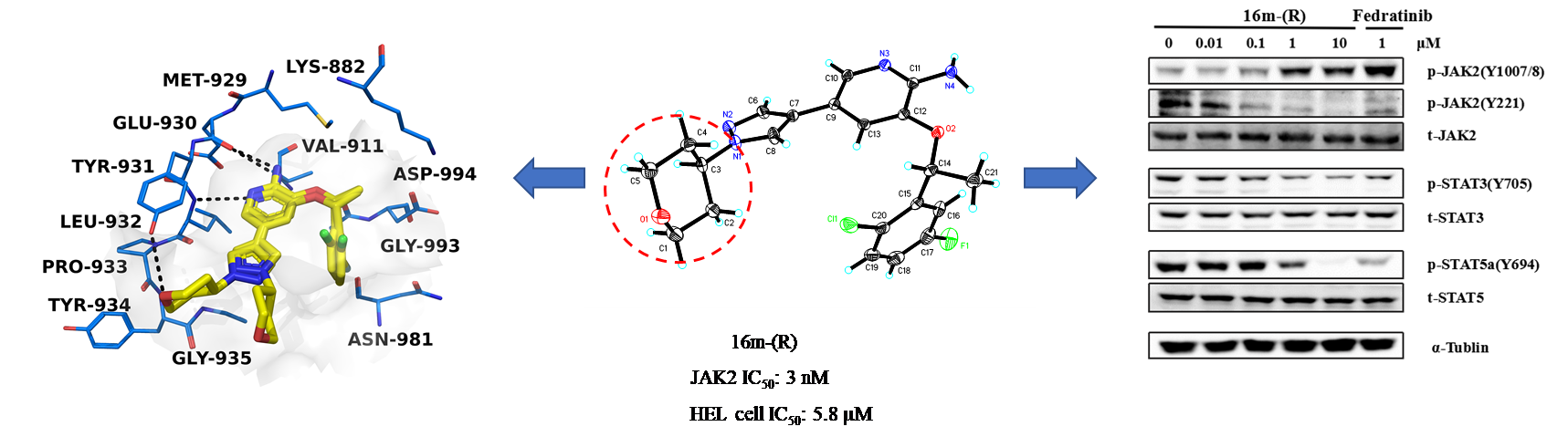 Janus kinases (JAKs) including JAK1, JAK2, JAK3, and TYK2 are members of a family of intracellular nonreceptor tyrosine kinases, which have been demonstrated to be critical in the cell signaling pathway and involved in inflammatory diseases and cancer. V617F mutation in JAK2 has been implicated in polycythaemia vera (PV), essential thrombocythaemia (ET) and myelofibroisis (MF). Here, we described the design, synthesis, and biological evaluation of a series of 2-aminopyridine derivatives. The results of enzymatic activity assays supported compound 16m-(R) as a potential and selective JAK2 inhibitor, which exhibited high inhibitory activity with an IC50 of 3 nM against JAK2, and 85- and 76-fold selectivity over JAK1 and JAK3, respectively. Structure-activity relationships (SAR) and mechanistic analysis demonstrated that 16m-(R) might be a promising selective JAK2 inhibitor for further study.
Janus kinases (JAKs) including JAK1, JAK2, JAK3, and TYK2 are members of a family of intracellular nonreceptor tyrosine kinases, which have been demonstrated to be critical in the cell signaling pathway and involved in inflammatory diseases and cancer. V617F mutation in JAK2 has been implicated in polycythaemia vera (PV), essential thrombocythaemia (ET) and myelofibroisis (MF). Here, we described the design, synthesis, and biological evaluation of a series of 2-aminopyridine derivatives. The results of enzymatic activity assays supported compound 16m-(R) as a potential and selective JAK2 inhibitor, which exhibited high inhibitory activity with an IC50 of 3 nM against JAK2, and 85- and 76-fold selectivity over JAK1 and JAK3, respectively. Structure-activity relationships (SAR) and mechanistic analysis demonstrated that 16m-(R) might be a promising selective JAK2 inhibitor for further study. Novel Selective Inhibitors of PfDHODH: Discovery and Optimization of Pyrimidone Derivatives
-
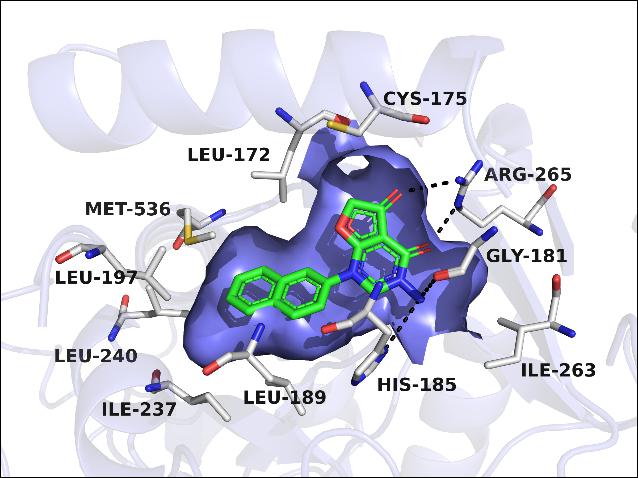 Inhibition of Plasmodium falciparum dihydroorotate dehydrogenase (PfDHODH) potentially represents a new treatment option for malaria, since P. falciparum relies entirely on de novo pyrimidine biosynthetic pathway for survival. In this work, a series of brand new scaffold PfDHODH-specific inhibitors pyrimidone derivatives were obtained from docking analyses and structural optimizations. The most potent inhibitor 26 showed excellent inhibitory activity against PfDHODH (IC50 = 23 nM) and high species selectivity over hDHODH. Through SAR studies, three preferential structural fragments were essential for these novel potent PfDHODH inhibitors: (1) bicyclic systems such as “naphthyl-like” substituents as the hydrophobic group; (2) two carbonyl group in the dihydrofuranone ring oriented in the same direction and formed hydrogen bonds with the polar residue (Arg265); (3) a hydrogen bond donor (NH2 in compound 20 and 26) was important for the inhibitory activity to interact with the imidazole group of His185. The results might be valuable for the novel scaffold PfDHODH inhibitors to be developed into new antimalarial agents.
Inhibition of Plasmodium falciparum dihydroorotate dehydrogenase (PfDHODH) potentially represents a new treatment option for malaria, since P. falciparum relies entirely on de novo pyrimidine biosynthetic pathway for survival. In this work, a series of brand new scaffold PfDHODH-specific inhibitors pyrimidone derivatives were obtained from docking analyses and structural optimizations. The most potent inhibitor 26 showed excellent inhibitory activity against PfDHODH (IC50 = 23 nM) and high species selectivity over hDHODH. Through SAR studies, three preferential structural fragments were essential for these novel potent PfDHODH inhibitors: (1) bicyclic systems such as “naphthyl-like” substituents as the hydrophobic group; (2) two carbonyl group in the dihydrofuranone ring oriented in the same direction and formed hydrogen bonds with the polar residue (Arg265); (3) a hydrogen bond donor (NH2 in compound 20 and 26) was important for the inhibitory activity to interact with the imidazole group of His185. The results might be valuable for the novel scaffold PfDHODH inhibitors to be developed into new antimalarial agents. Structure-Guided Design of Potent and Mutant-Selective EGFR L858R/T790M Inhibitors
-
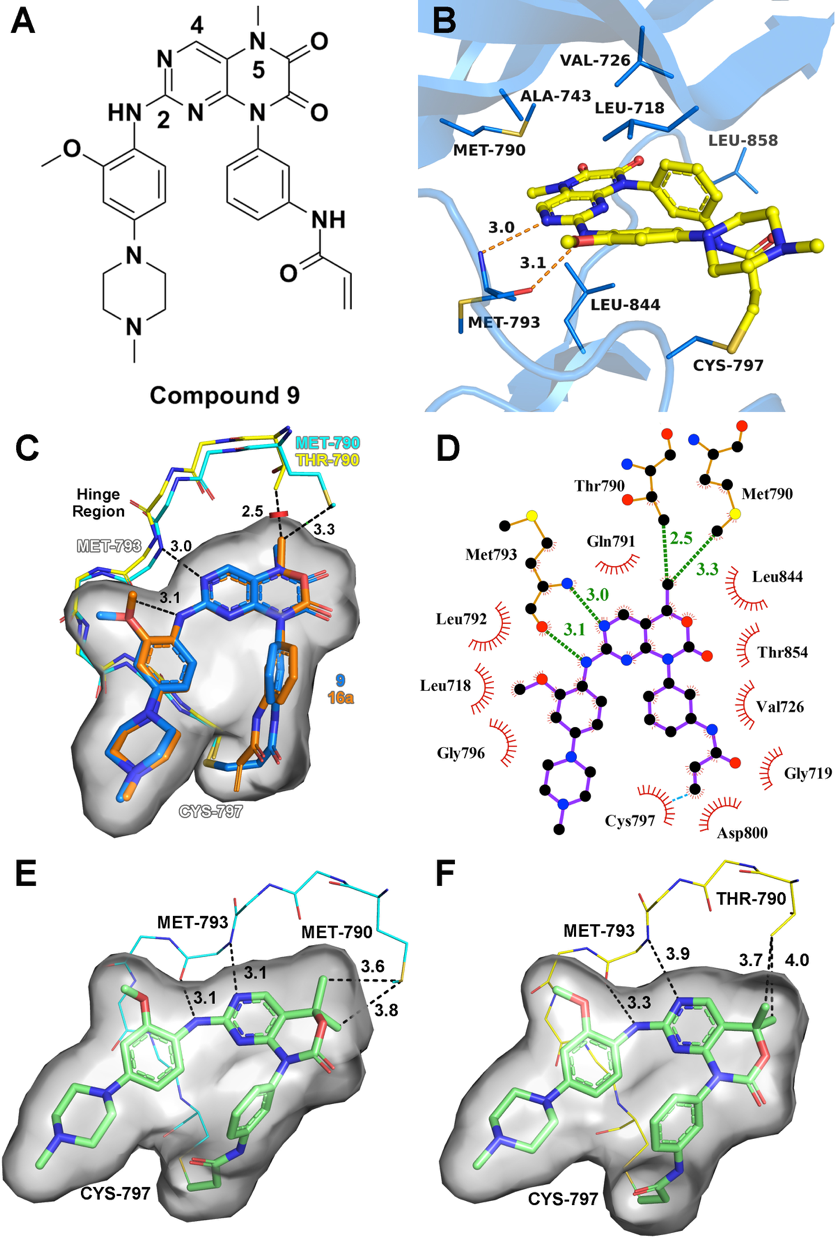 Epidermal growth factor receptor (EGFR) T790M acquired drug-resistance mutation has become a major clinical challenge for the therapy of non-small cell lung cancer. Here, we applied a structure-guided approach on the basis of the previous reported EGFR inhibitor (compound 9), and designed a series of C4-alkyl-1,4-dihydro-2H-pyrimido[4,5-d][1,3]oxazin-2-one derivatives as novel mutant-selective EGFR inhibitors. Finally, the most representative compound 20a was identified, which showed high selectivity at both enzymatic and cellular levels against EGFRL858R/T790M (H1975 cell lines) over EGFRWT (A431 cell lines). The representative compound 20a also showed promising antitumor efficiency in the in vivo antitumor efficacy study of H1975 xenograft mouse model driven by EGFRL858R/T790M. These results provide a new scaffold for the treatment of dual-mutant-driven non-small cell lung cancer.
Epidermal growth factor receptor (EGFR) T790M acquired drug-resistance mutation has become a major clinical challenge for the therapy of non-small cell lung cancer. Here, we applied a structure-guided approach on the basis of the previous reported EGFR inhibitor (compound 9), and designed a series of C4-alkyl-1,4-dihydro-2H-pyrimido[4,5-d][1,3]oxazin-2-one derivatives as novel mutant-selective EGFR inhibitors. Finally, the most representative compound 20a was identified, which showed high selectivity at both enzymatic and cellular levels against EGFRL858R/T790M (H1975 cell lines) over EGFRWT (A431 cell lines). The representative compound 20a also showed promising antitumor efficiency in the in vivo antitumor efficacy study of H1975 xenograft mouse model driven by EGFRL858R/T790M. These results provide a new scaffold for the treatment of dual-mutant-driven non-small cell lung cancer. Design of Potent and Selective EGFR Inhibitors against L858R/T790M Resistance Mutation
-
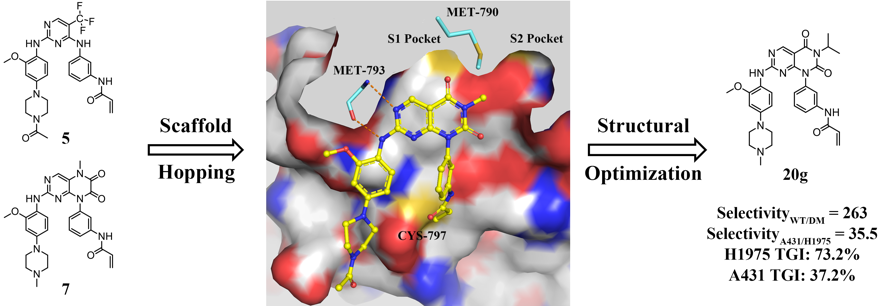 First-generation epidermal growth factor receptor (EGFR) inhibitors, gefitinib and erlotinib have achieved initially marked clinical efficacy for non-small cell lung cancer (NSCLC) patients with EGFR activating mutations. However, their clinical benefit was limited by the emergence of acquired resistance mutations. In most cases (approximately 60%), the resistance was caused by the secondary EGFR T790M gatekeeper mutation. Thus, it is still desirable to develop novel third-generation EGFR inhibitors to overcome T790M mutation while sparing wild-type (WT) EGFR. Herein, a series of pyrimido[4,5-d]pyrimidine-2,4(1H,3H)-dione derivatives were designed and synthesized. Among which, the most potent compound 20g not only demonstrated significant inhibitory activity and selectivity for EGFRL858R/T790M and H1975 cells in vitro, but also displayed outstanding antitumor efficiency in H1975 xenograft mouse model. The encouraging mutant-selective results at both in vitro and in vivo levels suggested that 20g might be used as a promising lead compound for further structural optimization as potent and selective EGFRL858R/T790M inhibitors.
First-generation epidermal growth factor receptor (EGFR) inhibitors, gefitinib and erlotinib have achieved initially marked clinical efficacy for non-small cell lung cancer (NSCLC) patients with EGFR activating mutations. However, their clinical benefit was limited by the emergence of acquired resistance mutations. In most cases (approximately 60%), the resistance was caused by the secondary EGFR T790M gatekeeper mutation. Thus, it is still desirable to develop novel third-generation EGFR inhibitors to overcome T790M mutation while sparing wild-type (WT) EGFR. Herein, a series of pyrimido[4,5-d]pyrimidine-2,4(1H,3H)-dione derivatives were designed and synthesized. Among which, the most potent compound 20g not only demonstrated significant inhibitory activity and selectivity for EGFRL858R/T790M and H1975 cells in vitro, but also displayed outstanding antitumor efficiency in H1975 xenograft mouse model. The encouraging mutant-selective results at both in vitro and in vivo levels suggested that 20g might be used as a promising lead compound for further structural optimization as potent and selective EGFRL858R/T790M inhibitors. Rational Design of Natural Product-derived Analogs as Novel and Long-acting DPP-4 Inhibitors
-
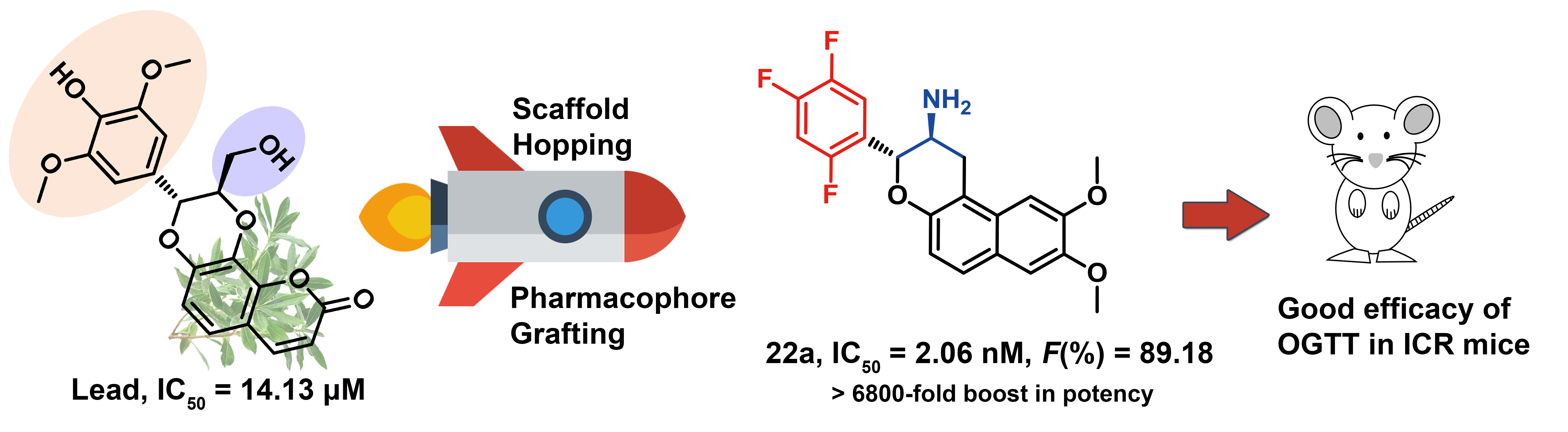 Starting from the lead isodaphnetin, a natural product inhibitor of DPP-4 discovered through a target fishing docking based approach, a series of novel 2-phenyl-3,4-dihydro-2H-benzo[f]chromen-3-amine derivatives as potent DPP-4 inhibitors are rationally designed utilizing highly efficient 3D molecular similarity based scaffold hopping as well as electrostatic complementary methods. Those ingenious drug design strategies bring us approximate 7400-fold boost in potency. Compounds 22a and 24a are the most potent ones (IC50 ≈ 2.0 nM) with good pharmacokinetic profiles. Compound 22a demonstrated stable pharmacological effect. A 3 mg/kg oral dose provided >80% inhibition of DPP-4 activity within 24 h, which is comparable to the performance of the long-acting control omarigliptin. Moreover, the efficacy of 22a in improving the glucose tolerance is also comparable with omarigliptin. In this study, not only promising DPP-4 inhibitors as long acting antidiabetic that are clinically on demand are identified, but the target fish docking and medicinal chemistry strategies were successfully implemented.
Starting from the lead isodaphnetin, a natural product inhibitor of DPP-4 discovered through a target fishing docking based approach, a series of novel 2-phenyl-3,4-dihydro-2H-benzo[f]chromen-3-amine derivatives as potent DPP-4 inhibitors are rationally designed utilizing highly efficient 3D molecular similarity based scaffold hopping as well as electrostatic complementary methods. Those ingenious drug design strategies bring us approximate 7400-fold boost in potency. Compounds 22a and 24a are the most potent ones (IC50 ≈ 2.0 nM) with good pharmacokinetic profiles. Compound 22a demonstrated stable pharmacological effect. A 3 mg/kg oral dose provided >80% inhibition of DPP-4 activity within 24 h, which is comparable to the performance of the long-acting control omarigliptin. Moreover, the efficacy of 22a in improving the glucose tolerance is also comparable with omarigliptin. In this study, not only promising DPP-4 inhibitors as long acting antidiabetic that are clinically on demand are identified, but the target fish docking and medicinal chemistry strategies were successfully implemented. Discovery of peptide inhibitors targeting hPD-1
-
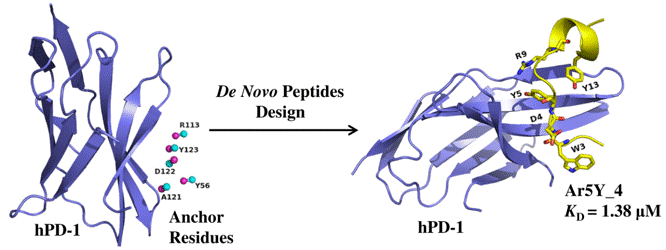 The hPD-1 is an important checkpoint receptors. Blocking the interaction of hPD-1 and its ligand hPD-L1 has been a promising immunotherapy in cancer treatment. we designed several hPD-1 binding peptides using a computational de novo peptide design method. The most potent peptide Ar5Y_4 showed a KD value of 1.38 ± 0.39 μM, comparable to the binding affinity of the cognate hPD-L1. A Surface Plasmon Resonance (SPR) competitive binding assay result indicated that Ar5Y_4 could inhibit the interaction of hPD-1/hPD-L1. Moreover, Ar5Y_4 could restore the function of Jurkat T cells which had been suppressed by stimulated HCT116 cells. So the peptides Ar5Y_4 can provide promising biologic candidates for cancer immunotherapy or diagnostics.
The hPD-1 is an important checkpoint receptors. Blocking the interaction of hPD-1 and its ligand hPD-L1 has been a promising immunotherapy in cancer treatment. we designed several hPD-1 binding peptides using a computational de novo peptide design method. The most potent peptide Ar5Y_4 showed a KD value of 1.38 ± 0.39 μM, comparable to the binding affinity of the cognate hPD-L1. A Surface Plasmon Resonance (SPR) competitive binding assay result indicated that Ar5Y_4 could inhibit the interaction of hPD-1/hPD-L1. Moreover, Ar5Y_4 could restore the function of Jurkat T cells which had been suppressed by stimulated HCT116 cells. So the peptides Ar5Y_4 can provide promising biologic candidates for cancer immunotherapy or diagnostics. Discovery of Selective EGFR Inhibitors against L858R/T790M Resistance Mutation
-
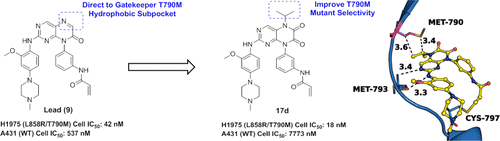 EGFR-targeted inhibitors (gefitinib and erlotinib) provided an effective strategy for the treatment of non-small-cell lung cancer. However, the EGFR T790M secondary mutation has become a leading cause of clinically acquired resistance to these agents. Herein, on the basis of the previously reported irreversible EGFR inhibitor (compound 9), we present a structure-based design approach, which is rationalized via analyzing its binding model and comparing the differences of gatekeeper pocket between the T790M mutant and wild-type (WT) EGFR kinases. Guided by these results, a novel 6,7-dioxo-6,7-dihydropteridine scaffold was discovered and hydrophobic modifications at N5-position were conducted to strengthen nonpolar contacts and improve mutant selectivity over EGFRWT. Finally, the most representative compound 17d was identified. This work demonstrates the power of structure-based strategy in discovering lead compounds and provides molecular insights into the selectivity of EGFRL858R/T790M over EGFRWT, which may play an important role in designing new classes of mutant-selective EGFR inhibitors.
EGFR-targeted inhibitors (gefitinib and erlotinib) provided an effective strategy for the treatment of non-small-cell lung cancer. However, the EGFR T790M secondary mutation has become a leading cause of clinically acquired resistance to these agents. Herein, on the basis of the previously reported irreversible EGFR inhibitor (compound 9), we present a structure-based design approach, which is rationalized via analyzing its binding model and comparing the differences of gatekeeper pocket between the T790M mutant and wild-type (WT) EGFR kinases. Guided by these results, a novel 6,7-dioxo-6,7-dihydropteridine scaffold was discovered and hydrophobic modifications at N5-position were conducted to strengthen nonpolar contacts and improve mutant selectivity over EGFRWT. Finally, the most representative compound 17d was identified. This work demonstrates the power of structure-based strategy in discovering lead compounds and provides molecular insights into the selectivity of EGFRL858R/T790M over EGFRWT, which may play an important role in designing new classes of mutant-selective EGFR inhibitors. Identification of novel CAs inhibitors from the chemical database
-
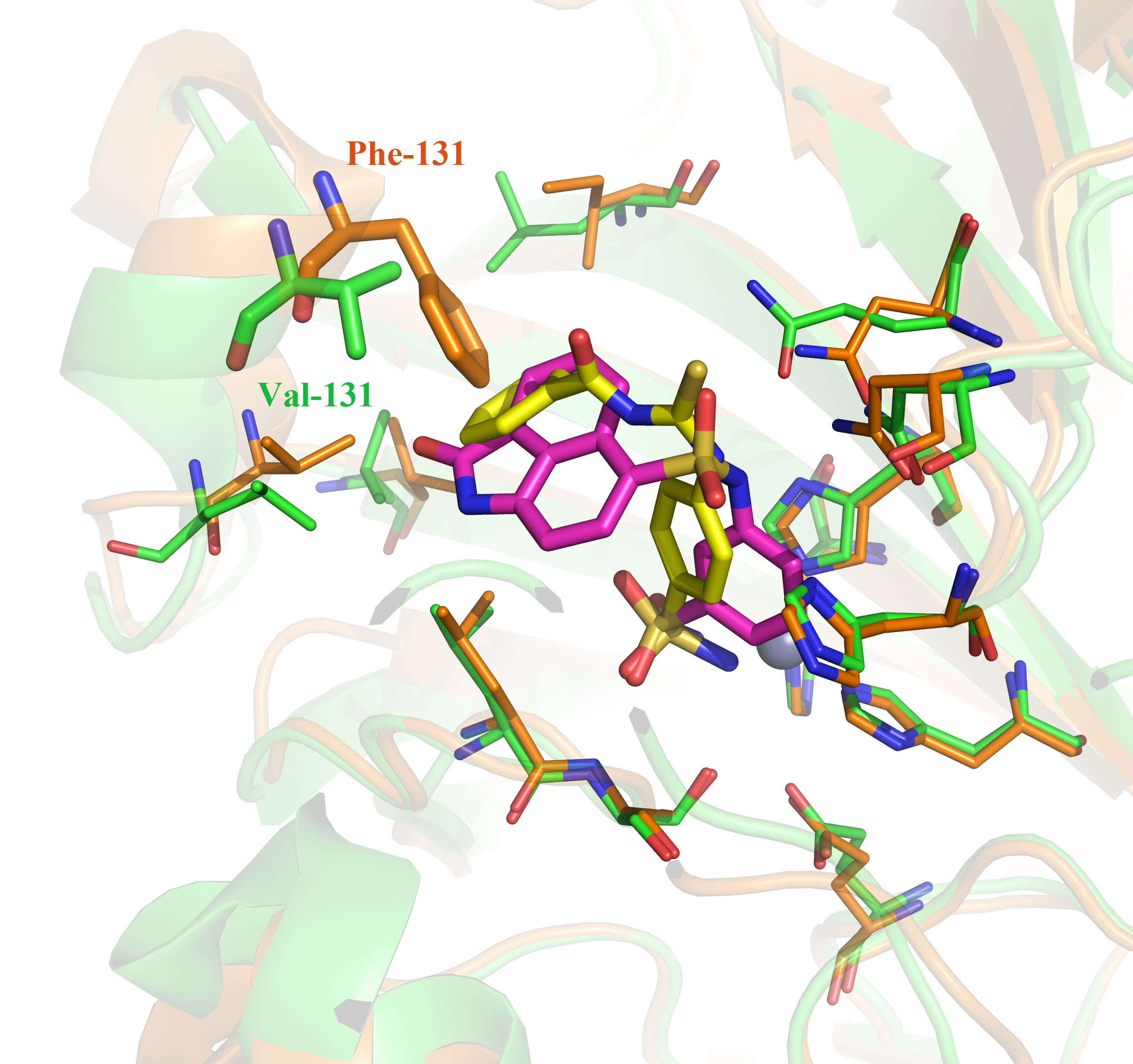 Through the structure-based virtual screening, a new series of benzene sulfonamides are identified as potent inhibitors of Carbonic Anhydrase (CA) IX. Not only two most potent compounds 1 and 2 (the IC50 against CA IX are 2.86 and 3.11 nM respectively) are identified, but also two compounds 1 and 9 show high selectivity for tumor-target CA IX over the CA II (the selectivity ratios are 21.3 and 136.6 respectively). According to the docking analysis, the selectivity might be caused by the ligands with big function groups clashed with the Phe131 of CA II but not with the Val131 of CA IX.
Through the structure-based virtual screening, a new series of benzene sulfonamides are identified as potent inhibitors of Carbonic Anhydrase (CA) IX. Not only two most potent compounds 1 and 2 (the IC50 against CA IX are 2.86 and 3.11 nM respectively) are identified, but also two compounds 1 and 9 show high selectivity for tumor-target CA IX over the CA II (the selectivity ratios are 21.3 and 136.6 respectively). According to the docking analysis, the selectivity might be caused by the ligands with big function groups clashed with the Phe131 of CA II but not with the Val131 of CA IX.
Bioinformatics
DNA-binding proteins prediction using SVM and comprehensive feature analysis
-
 In this work, the focus is how to transform these informative features into uniform numeric representation appropriately and improve the prediction accuracy of our SVM-based classifier for DNA-BPs. A systematic representation of some selected features known to perform well is investigated here. Firstly, four kinds of protein properties are obtained and used to describe the protein sequence. Secondly, three different feature transformation methods (OCTD, AC and SAA) are adopted to obtain numeric feature vectors from three main levels: Global, Nonlocal and Local of protein sequence and their performances are exhaustively investigated. At last, the mRMR-IFS feature selection method and ensemble learning approach are utilized to determine the best prediction model.
In this work, the focus is how to transform these informative features into uniform numeric representation appropriately and improve the prediction accuracy of our SVM-based classifier for DNA-BPs. A systematic representation of some selected features known to perform well is investigated here. Firstly, four kinds of protein properties are obtained and used to describe the protein sequence. Secondly, three different feature transformation methods (OCTD, AC and SAA) are adopted to obtain numeric feature vectors from three main levels: Global, Nonlocal and Local of protein sequence and their performances are exhaustively investigated. At last, the mRMR-IFS feature selection method and ensemble learning approach are utilized to determine the best prediction model. Accurate prediction of RNA-binding protein residues with two discriminative structural descriptors
-
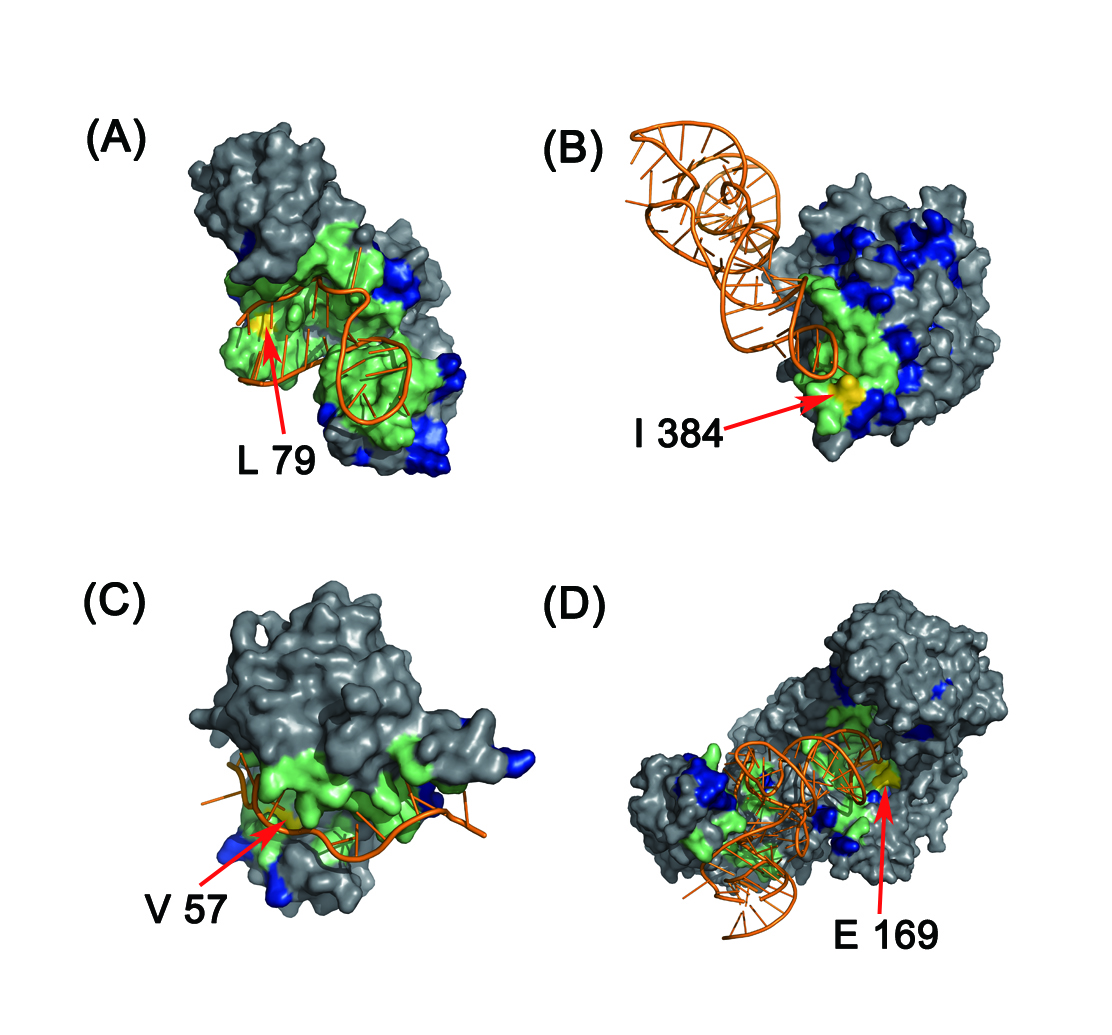 In this study, we designed two discriminative structure-derived features, namely residue electrostatic surface potential and triplet interface propensity, to characterize a protein residue together with other commonly used descriptors. A comprehensive analysis of the two newly designed features from different aspects demonstrated that the two features have excellent discriminative power on a large dataset and may reflect the underlying mechanisms of RNA-protein interactions. To incorporate information from neighbor residues to determine the RNA-binding properties of each target residue, the optimal patch type and patch size for different features are searched, and by using the searched optimal patch type and patch size for each used feature, a random forest classifier is developed and implemented in the web server RNAProSite. From the results of a fivefold cross validation on a training set and the prediction performance on a test set, we concluded that our method can predict RBRs with results better than or comparable to those of the existing approaches and could assist researchers in performing more targeted assays.
In this study, we designed two discriminative structure-derived features, namely residue electrostatic surface potential and triplet interface propensity, to characterize a protein residue together with other commonly used descriptors. A comprehensive analysis of the two newly designed features from different aspects demonstrated that the two features have excellent discriminative power on a large dataset and may reflect the underlying mechanisms of RNA-protein interactions. To incorporate information from neighbor residues to determine the RNA-binding properties of each target residue, the optimal patch type and patch size for different features are searched, and by using the searched optimal patch type and patch size for each used feature, a random forest classifier is developed and implemented in the web server RNAProSite. From the results of a fivefold cross validation on a training set and the prediction performance on a test set, we concluded that our method can predict RBRs with results better than or comparable to those of the existing approaches and could assist researchers in performing more targeted assays.
Methodology
Statistical Analysis, Investigation, and Prediction of the Water Positions
-
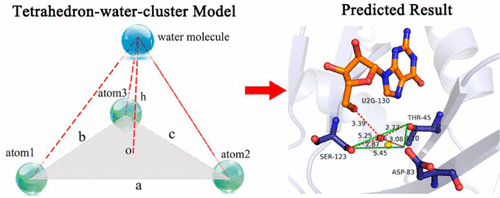 Water molecules play a crucial role in biomolecular associations by mediating a hydrogen bond network or filling spaces with van der Waals interactions. Although current drug design technologies have taken water molecule interactions into account, their applications are still limited to their reliance on either excessive computer resources or a particular potential energy model. Here, we introduce a statistical method that is based on experimentally determined water molecules in the binding sites of high-resolution X-ray crystal structures to predict the potential hydration sites in the binding sites of crystal structures of interest. By clustering and analyzing the various interaction patterns of water molecules in the training data set, we derived a tetrahedron-water-cluster model based on a series of residue group triplets that form feature triangles of different shapes. In the tetrahedral-water-cluster model, a triplet of three polar atoms in the residue group triplet acts as the vertices of the bottom triangle of the tetrahedron, and the water molecule that interacts with these three polar atoms is set as the top vertex of the tetrahedron. By comparing the shapes of the bottom triangles in the training data set with the shape of the triangle in the residue group triplets in the crystal structure of interest, we can identify the bottom triangle that is most similar to the one in the residue group triplet of the crystal structure of interest. According to the tetrahedron-water-cluster model, the hydration site for the residue group triplet in the crystal structure of interest can be predicted based on the height of the tetrahedron that has the most similar bottom triangle in the training data set. A test set containing 193 crystal structures was used to evaluate model performance, and extensive comparison with the recently published program Dowser++ revealed that our model is at least as good at providing an accurate set of the potential hydration sites in crystal structures of interest.
Water molecules play a crucial role in biomolecular associations by mediating a hydrogen bond network or filling spaces with van der Waals interactions. Although current drug design technologies have taken water molecule interactions into account, their applications are still limited to their reliance on either excessive computer resources or a particular potential energy model. Here, we introduce a statistical method that is based on experimentally determined water molecules in the binding sites of high-resolution X-ray crystal structures to predict the potential hydration sites in the binding sites of crystal structures of interest. By clustering and analyzing the various interaction patterns of water molecules in the training data set, we derived a tetrahedron-water-cluster model based on a series of residue group triplets that form feature triangles of different shapes. In the tetrahedral-water-cluster model, a triplet of three polar atoms in the residue group triplet acts as the vertices of the bottom triangle of the tetrahedron, and the water molecule that interacts with these three polar atoms is set as the top vertex of the tetrahedron. By comparing the shapes of the bottom triangles in the training data set with the shape of the triangle in the residue group triplets in the crystal structure of interest, we can identify the bottom triangle that is most similar to the one in the residue group triplet of the crystal structure of interest. According to the tetrahedron-water-cluster model, the hydration site for the residue group triplet in the crystal structure of interest can be predicted based on the height of the tetrahedron that has the most similar bottom triangle in the training data set. A test set containing 193 crystal structures was used to evaluate model performance, and extensive comparison with the recently published program Dowser++ revealed that our model is at least as good at providing an accurate set of the potential hydration sites in crystal structures of interest. Multi-Body Interactions in Molecular Docking Program Devised with Key Water Molecules
-
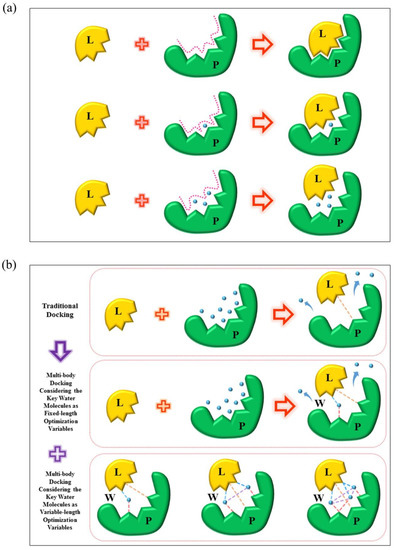 Water molecules play an important role in modeling protein-ligand interactions. However, traditional molecular docking methods often ignore the impact of the water molecules by removing them without any analysis or keeping them as a static part of the proteins or the ligands. Hence, the accuracy of the docking simulations will inevitably be damaged. Here, we introduce a multi-body docking program which incorporates the fixed or the variable number of the key water molecules in protein-ligand docking simulations. The program employed NSGA II, a multi-objective optimization algorithm, to identify the binding poses of the ligand and the key water molecules for a protein. To this end, a force-field-based hydration-specific scoring function was designed to favor estimate the binding affinity considering the key water molecules. The program was evaluated in aspects of the docking accuracy, cross-docking accuracy, and screening efficiency. When the numbers of the key water molecules were treated as fixed-length optimization variables, the docking accuracy of the multi-body docking program achieved a success rate of 80.58% for the best RMSD values for the recruit of the ligands smaller than 2.0 Å. The cross-docking accuracy was investigated on the presence and absence of the key water molecules by four protein targets. The screening efficiency was assessed against those protein targets. Results indicated that the proposed multi-body docking program was with good performance compared with the other programs. On the other side, when the numbers of the key water molecules were treated as variable-length optimization variables, the program obtained comparative performance under the same three evaluation criterions. These results indicated that the multi-body docking with the variable numbers of the water molecules was also efficient. Above all, the multi-body docking program developed in this study was capable of dealing with the problem of the water molecules that explicitly participating in protein-ligand binding.
Water molecules play an important role in modeling protein-ligand interactions. However, traditional molecular docking methods often ignore the impact of the water molecules by removing them without any analysis or keeping them as a static part of the proteins or the ligands. Hence, the accuracy of the docking simulations will inevitably be damaged. Here, we introduce a multi-body docking program which incorporates the fixed or the variable number of the key water molecules in protein-ligand docking simulations. The program employed NSGA II, a multi-objective optimization algorithm, to identify the binding poses of the ligand and the key water molecules for a protein. To this end, a force-field-based hydration-specific scoring function was designed to favor estimate the binding affinity considering the key water molecules. The program was evaluated in aspects of the docking accuracy, cross-docking accuracy, and screening efficiency. When the numbers of the key water molecules were treated as fixed-length optimization variables, the docking accuracy of the multi-body docking program achieved a success rate of 80.58% for the best RMSD values for the recruit of the ligands smaller than 2.0 Å. The cross-docking accuracy was investigated on the presence and absence of the key water molecules by four protein targets. The screening efficiency was assessed against those protein targets. Results indicated that the proposed multi-body docking program was with good performance compared with the other programs. On the other side, when the numbers of the key water molecules were treated as variable-length optimization variables, the program obtained comparative performance under the same three evaluation criterions. These results indicated that the multi-body docking with the variable numbers of the water molecules was also efficient. Above all, the multi-body docking program developed in this study was capable of dealing with the problem of the water molecules that explicitly participating in protein-ligand binding.
Natural Products
Identification of novel MMPs inhibitors from the natrual product database
-
 19 natural compounds with diverse structures are identified as potential MMPIs using structure-based virtual screening from the 4000 natural products. Hydroxycinnamic acid or similarity groups of natural products are important for potent inhibitory and selectivity against MMPs, and the solvent effect in the S1’ pocket can effect the hydrophobic interactions and hydrogen bonds between MMPIs and MMPS, which make MMPIs show certain selectivity for a specific MMP enzyme. Furthermore, compound 5 can reduces the expression of both MMP-2 and active-MMP-9, and suppress the migration of MDA-MB-231 in a wound healing assay, which may be further developed as anticancer agent
19 natural compounds with diverse structures are identified as potential MMPIs using structure-based virtual screening from the 4000 natural products. Hydroxycinnamic acid or similarity groups of natural products are important for potent inhibitory and selectivity against MMPs, and the solvent effect in the S1’ pocket can effect the hydrophobic interactions and hydrogen bonds between MMPIs and MMPS, which make MMPIs show certain selectivity for a specific MMP enzyme. Furthermore, compound 5 can reduces the expression of both MMP-2 and active-MMP-9, and suppress the migration of MDA-MB-231 in a wound healing assay, which may be further developed as anticancer agent Jatropholane-Type Diterpenes fromEuphorbia sikkimensis
-
 Four new jatropholane-type diterpenes (1−4), named sikkimenoids A−D, were isolated from the aerial parts ofEuphorbia sikkimensis. The structural elucidations of 1−4 were accomplished by extensive NMR analyses, and their absolute configurations were established by ECD calculations. Compound2exhibited weak antiangiogenic activity with an IC50 value of 43.0μM when evaluated using a zebrafish model.
Four new jatropholane-type diterpenes (1−4), named sikkimenoids A−D, were isolated from the aerial parts ofEuphorbia sikkimensis. The structural elucidations of 1−4 were accomplished by extensive NMR analyses, and their absolute configurations were established by ECD calculations. Compound2exhibited weak antiangiogenic activity with an IC50 value of 43.0μM when evaluated using a zebrafish model.
Web Server
iDrug: a web-accessible and interactive drug discovery and design platform
-
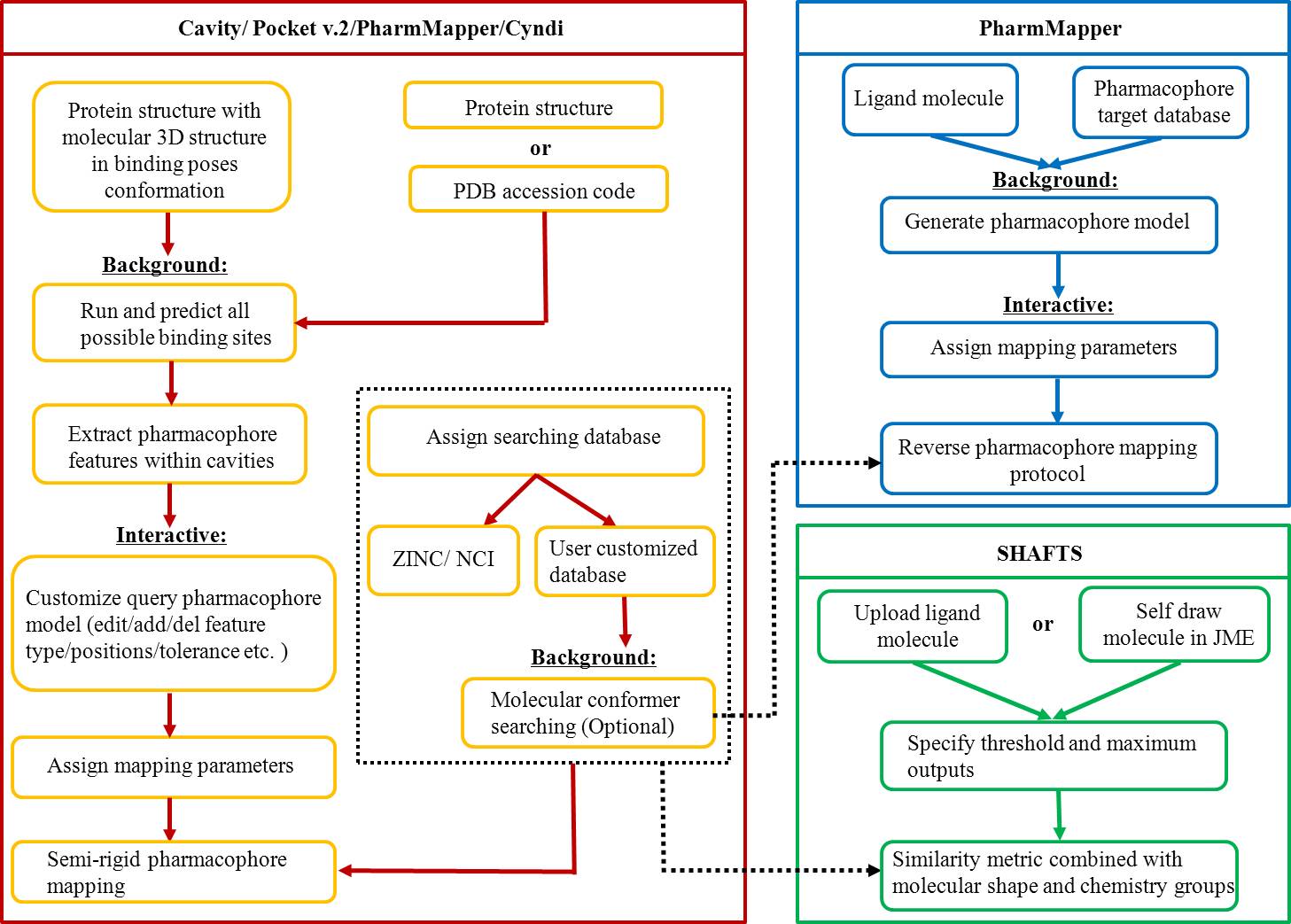 We presented a versatile, user-friendly, and efficient online tool for computer-aided drug design based on pharmacophore and 3D molecular similarity searching. The web interface enables binding sites detection, virtual screening hits identification, and drug targets prediction in an interactive manner through a seamless interface to all adapted packages (e.g., Cavity, PocketV.2, PharmMapper, SHAFTS). Several commercially available compound databases for hit identification and a well-annotated pharmacophore database for drug targets prediction were integrated in iDrug as well.
We presented a versatile, user-friendly, and efficient online tool for computer-aided drug design based on pharmacophore and 3D molecular similarity searching. The web interface enables binding sites detection, virtual screening hits identification, and drug targets prediction in an interactive manner through a seamless interface to all adapted packages (e.g., Cavity, PocketV.2, PharmMapper, SHAFTS). Several commercially available compound databases for hit identification and a well-annotated pharmacophore database for drug targets prediction were integrated in iDrug as well.
User Login
Web Services
Contact us
Honglin Li's Lab
Shanghai Key Laboratory of New Drug Design
School of Pharmacy
East China University of Sci. & Tech.
Room 527, Building 18, 130 Meilong Road,
Shanghai, 200237, P. R. China
Tel: (86) 21 6425 0213 Prof. Honglin Li
hlli@ecust.edu.cn
Copyright © 2026 Prof. HongLin Li's Group, School of Pharmacy, East China University of Science & Technology · All Right Reserved.
沪ICP备19004698号-1 |  沪公网安备31011302004713号
沪公网安备31011302004713号