Workflow Overview
Step 1: Input molecule and conditions
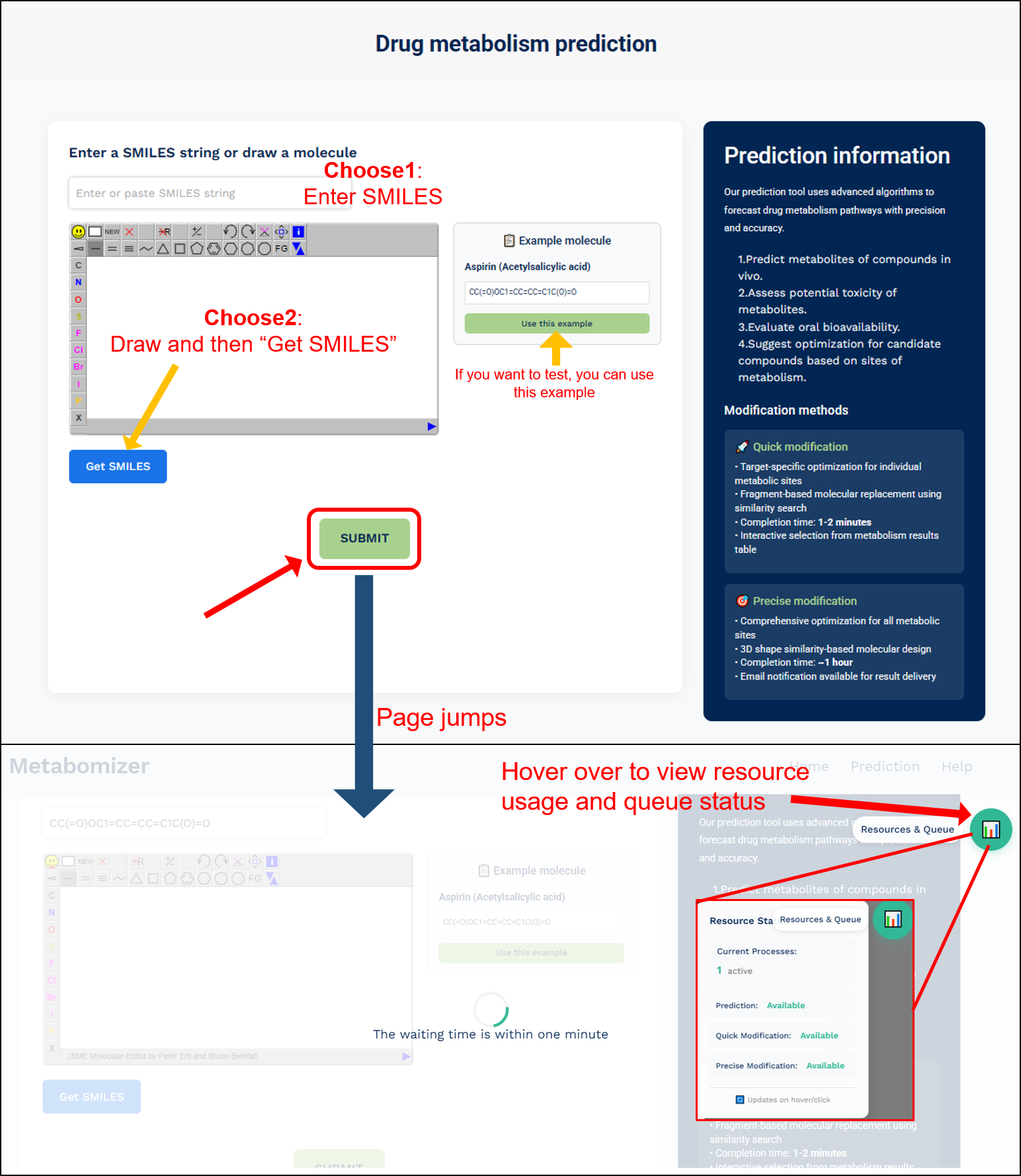
Prepare a SMILES string or chemical structure for the compound of interest. When you enter the 'Prediction' page, you can process the prediction step.
CRITICAL STEP: There are 2 ways to choose for input molecule:
Choose 1: Enter SMILES directly
- Enter or paste a valid SMILES string into the input box at the top
- Supports both canonical and noncanonical SMILES formats
- Ensure the SMILES string is properly formatted without any errors
Choose 2: Draw the molecule
- Use the built-in molecular drawing tool to construct your molecule
- Click the "Get SMILES" button after drawing to convert your structure to SMILES format
- The converted SMILES will automatically appear in the input box
Additional fatures:
- Example Molecule: If you want to test the system, you can use the provided example (Aspirin/Acetylsalicylic acid) by clicking the "Use this example" button
- Submit: After entering or drawing your molecule, click the "SUBMIT" button to start the prediction process
- Page Navigation: The page will automatically jump to show the calculation progress
- Resource Monitor: Hover over the resource icon in the top right corner to view resource usage and queue status
Important notes:
- Remove any salts or solvent molecules from your structure before submission
- Verify that your SMILES representation is valid and error-free
- Do not navigate away from the page while the calculation is running
- The waiting time is typically within one minute for standard predictions
- You can monitor the calculation progress and resource availability through the status indicators
Step 2: Result view and modification options
Once the calculation is complete, the results page will display comprehensive information about your molecule in three main sections:
1. Input molecule properties:
- Input SMILES string and 2D molecular structure visualization
- Detailed molecular properties including:
- Physicochemical parameters (cLogP, MW, TPSA, etc.)
- Structural features (nHet, nRing, HBD, HBA)
- Predicted properties (Toxicity status, Bioavailability percentage)
- Color-coded metabolic probability indicators
2. Metabolism of input molecule:
- Tabulated results showing predicted metabolites with:
- Rank ordering of metabolites
- 2D structures of metabolites
- SMILES representations
- Reaction templates (with "More info" button for detailed reaction information)
- Reaction types
- Toxicity predictions
- "Modification" options for each metabolic site
3. Modification Options:
- Quick modification:
- Available through the "Modification" button in the metabolite table
- Targets specific metabolic sites you are interested in
- Processing time: 1-2 minutes
- Precise modification:
- Accessible via the "Click for modifying all metabolic sites using 3D shape similarity" button
- Comprehensive optimization of all metabolic sites
- Processing time: approximately 1 hour
- Results delivered via email (optional)
- Generates a unique results URL for accessing the modifications
Additional features:
- Detailed reaction information available through "More info" buttons
- Reference links to source databases where available
- Progress indicators for both quick and precise modification processes

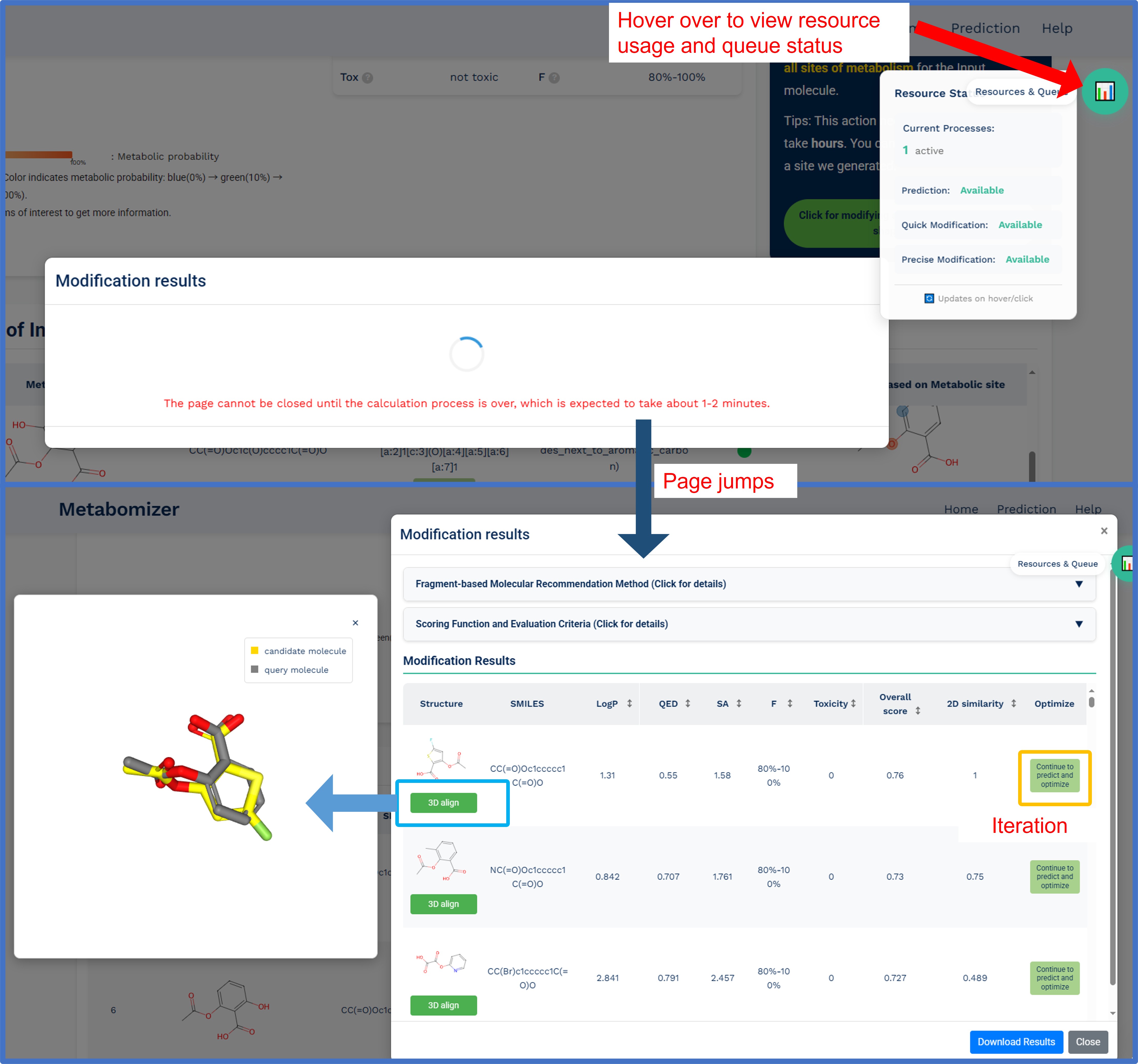
Step 3: Modification results and analysis
After selecting and running the modification process, the system will display comprehensive optimization results in an interactive interface:
Fragment-based molecular recommendation process:
- Identification of fragments associated with the molecule's metabolic sites
- Fragment replacement and linkage optimization
- Conformational screening and validation
- Ranking using multi-criteria evaluation functions
Results display features:
- Interactive results table:
- 2D molecular structures of optimized compounds
- SMILES strings for each recommended molecule
- Comprehensive scoring metrics (LogP, QED, SA, F, Toxicity)
- Overall optimization score and 2D similarity to parent molecule
- "Optimize" button for iterative refinement
- 3D molecular visualization:
- Interactive 3D alignment viewer showing query and candidate molecules
- Real-time comparison of original and modified structures
- 3D shape similarity assessment