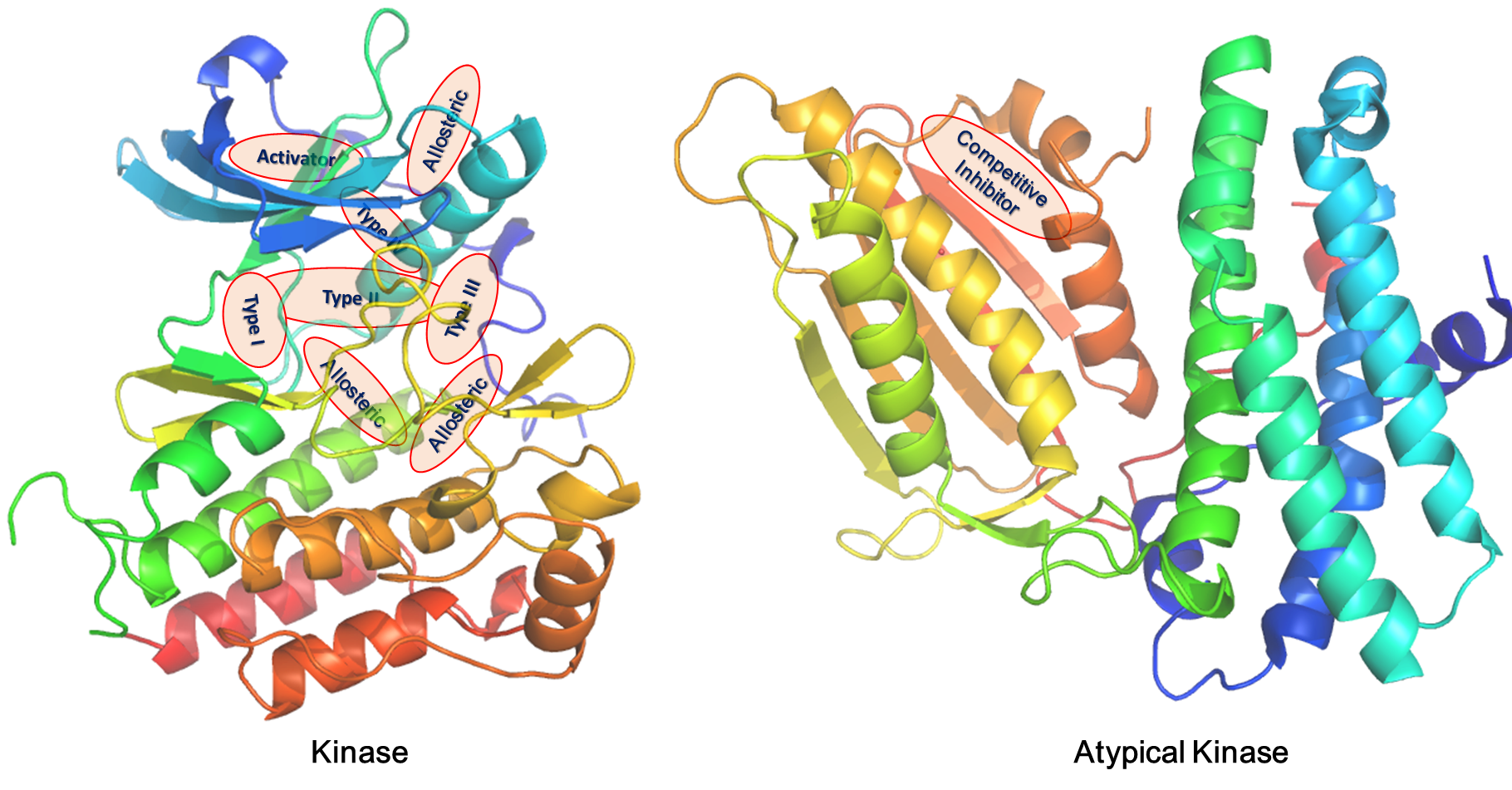
The kinase structural database KinLigDB currently contains 4,219 curated kinase-ligand complex structures of 297 human kinases (covering 106 kinase families); most of them are related to human diseases. About 75% of these kinases have at least two structures with different ligands, and 92 kinases have ≥10 complex structures. A lot of 396 binding sites were found and defined for the kinases in the database. Notably, 73 kinases have two or more different binding sites. The analyses of the binding modes identified eight types of ligands, including type I inhibitor (3167 entries), type II inhibitor (283 entries), type III inhibitor (23 entries), type IV inhibitor (9 entries), competitive inhibitor (554 entries), covalent inhibitor (31 entries), activator (49 entries), and allosteric ligand (103 entries). A total of 2805 kinase-ligand complexes were annotated with the experimental binding data.