iDrug User Manual
-
1. Introduction
- The iDrug User Manual is intended to help you perform protein binding sites detection, pharmacophore searching, potential target candidates identification and 3D similarity calculation with iDrug.
-
1.1 Running iDrug Server
Requirements: Best view with IE 8.0+, Firefox, Chrome, Safari, Opera. If you are unable to open the Jmol applet, please manually change the java security level to medium from high in the Java Control Panel and reload the page.
Three possible ways are provided to start with iDrug.- Register and Login (recommended, but not compulsory): You could first fill some brief register information to create your own account. Please do provide a valid e-mail address so as to receive the activation mail for using iDrug as well as receive the mail reminder about the job status.
- Guest: You could directly have access to iDrug as a Guest.
- Load session: Upload the session file to continue the last updated tasks.
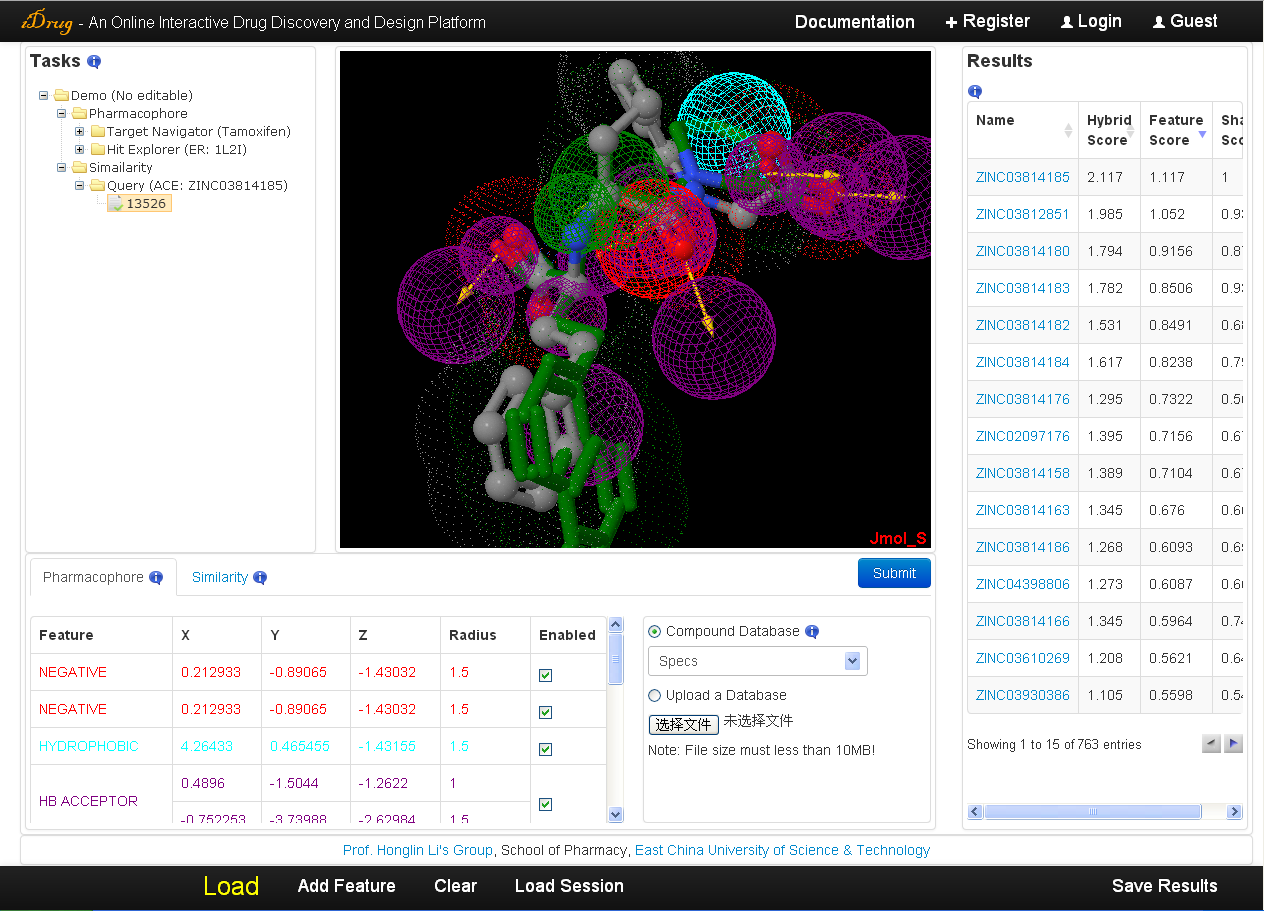
1.2 iDrug Panels
This section describes the five main panels of iDrug.
In the left part of the interface is the user task panel. Once the user logon to iDrug and perform the tasks, then any job will be added to the task directory bounded to the user account. The user can check the individual result by clicking on the corresponding job ID, which is returned by the server. Moreover, a simple tip is displayed when a job is created or completed, and each job can be renamed or deleted.
In the middle part of the interface is a Java-based Jmol molecular viewer.
In the right part of the interface are the results of specific tasks performed by iDrug. The implication of this result table is described in detail in Section
In the bottom left of the interface is the query parameter table. Two modules, namely "Pharmacophore" and "similarity" are provided.- The pharmacophore query editor supports the interactive modification of the properties of the pharmacophore, including the type (hydrophobic, hydrogen bond acceptor/donor, negative/positive centers or aromatic) of the pharmacophore features, position information, and an enabled/disabled option. A list of chemical searching databases in the drop-down box or a local mini-database in mol2 format can be selected as the target library.
- The similarity query editor contains the searching database selection (the same as pharmacophore query editor), the maximum number of hit compounds, and the similarity threshold scaled to a value between 0 and 2 since iDrug users the hybrid approach for 3D molecular similarity calculation.
In the bottom part of the interface, the following buttons appear:- Load----Opens the Load Files dialog box, in which the user can upload files and submit jobs. The general description is given in Section
- Add Feature----Add a new pharmacophore feature (hydrophobic by default) in the pharmacophore query editor, which is also shown as sphere in Jmol.
- Clear----Clear all the content in Jmol, result table, and query parameter table.
- Save Results----Save the calculation results returned by iDrug. The detailed information is given in Section
1.3 Using iDrug
The first steps of a typical job might proceed using the iDrug panels as follows:- Uploading the query molecules through the "Load" button.
- Creating a valid pharmacophore query by using the "Add Feature" button. Three at least pharmacophore features must be provided, since iDrug using the triangle hashing mapping technology.
- Resubmit jobs based on the former tasks in the task management panel.
1.4 Submit Job
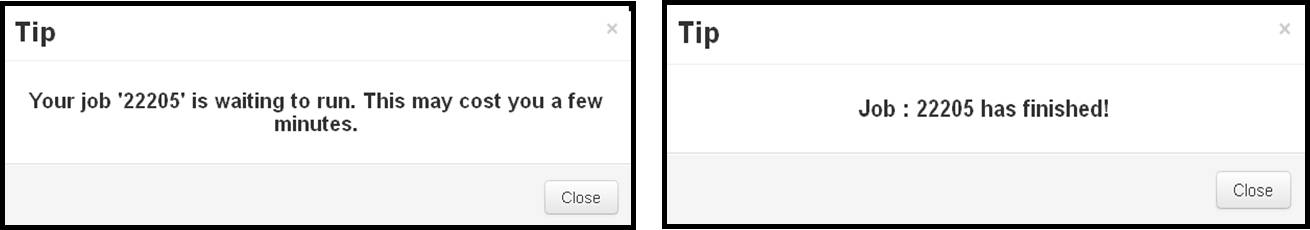
- Just click on the "submit" button (either in the "Load Panel" or in the bottom of the page) and iDrug will show tips on which the unique job id assigned as well as the propable computing time will be displayed. The job status is self-refreshed per 5 minutes and will show tips to users as soon as the job finished.
- Due to the limit of server load, the submitted jobs are pushed into the queue sequentially before running. The users are notified with the queue status as well as get access to the final results in the user task panel.
-
2. Protein Binding Sites Detection
- iDrug offers a ligand-binding site of the target protein analysis facility using Cavity, a purely geometrical method to find potential pockets. A receptor-based pharmacophore model is also suggested by Cavity.
-
2.1 Query molecule preparation
iDrug requires a protein and a ligand (optional) as the query, the user may clicking on the "Load" button and select the "Pharmacophore" work type. In the "Hit Explorer" frame, three possible ways are provided:
- Provide a simple standard protein by uploading a file in PDB format.
- Input the PDB accession code which is available in Protein Data Bank.
- Provide a receptor file in PDB format with a molecular structure by uploading a file in mol2 format. In this way, iDrug will only detect around the given ligand file.
After uploading the desired files, clicking on the "Submit" button and the server will pop-up a dialog box like "Your job 13207 is running now."
2.2 Results visualization
When the server popup a dialog box like "Job: 13027 is completed!", then the user can click on the corresponding job ID in the task tree to view the returned results.
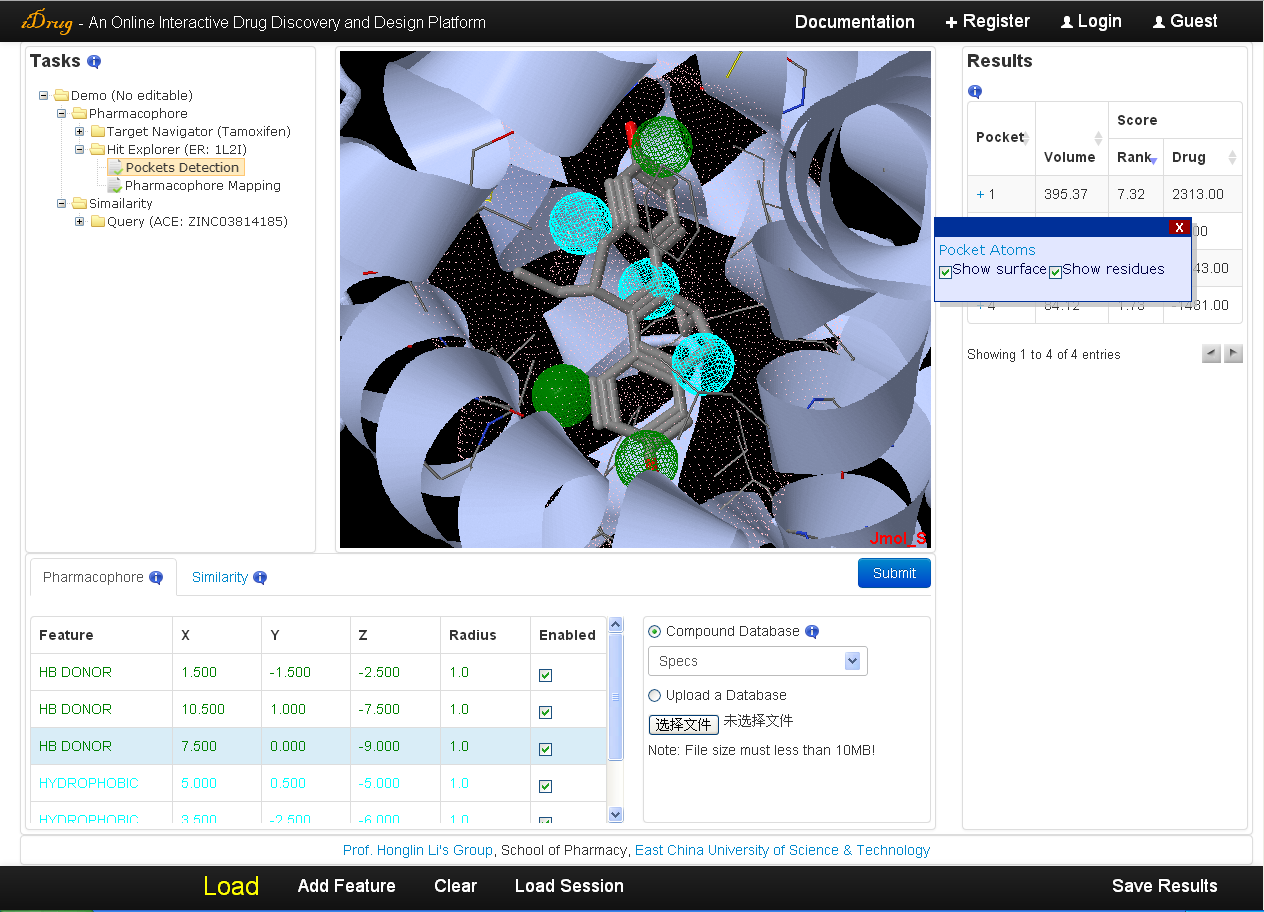
iDrug outputs all the potential binding sites of the protein ordered by a ranking score evaluated by the volume, surface area, hydrogen bond, and hydrophobic properties of the pockets. The detailed information is as follows:- In the result table, the user can also re-rank the result list by the volume of the pockets or druggability score via clicking the corresponding column.
- A pop-up window will appear by clicking the "+" mark in the "Pocket" column, which illustrates the detailed information of each binding site candidate, including a hyperlink to a list of atoms forming the pocket, the surface shape and the residues of the binding site with checkboxes allowing the user to show/hide the 3D chemical structures in Jmol applet.In the result table, the user can also re-rank the result list by the volume of the pockets or druggability score via clicking the corresponding column.
- The pharmacophore model derived inside the binding site is also shown in the pharmacophore query editor and Jmol applet when the user clicking the "+" mark.
- All the pockets information, including the surface shape, the volume shape, the residues, the key interaction site and pharmacophore model in PDB format, as well as pocket atoms in TXT format prefixed with the binding-site number are downloadable via the "Save Results" button.
NOTE: iDrug will take 2-5 minutes to perform cavity detection and receptor-based pharmacophore modeling!
-
3. Pharmacophore Search
- iDrug maps the query pharmacophore with the compounds of chemical database using triangle hashing method.
-
3.1 Pharmacophore query refinement
The user can change the type of each pharmacophore feature, the Cartesian coordinates, and the tolerance radius by double-clicking the corresponding frame. In addition, when the user click the relevant row, the mesh sphere shown in Jmol will be filled so as to distinguish individual features. Two possible ways are provided by iDrug to start editing the pharmacophore query:- Editing the pharmacophore model derived from the protein binding site (see in Section 2).
- Creating pharmacophore model through "Add Feature" button.
3.2 Searching database preparation
iDrug offers over 800,000 purchasable compounds of Specs, MayBridge, ZINC Leadlike Set, and NCI Open Database to perform pharmacophore-based virtual screening. Since iDrug uses semi-flexible alignment strategy, a conformer ensemble is generated by in-house program Cyndi with each compound having maximum 50 low-energy conformations.
A mini-database in mol2 format with the maximum size less than 10MB uploaded by the users as the target for screening is also allowed by iDrug.
3.3 Mapping parameters customization
After editing the pharmacophore query and selecting the searching database, the user may just click on the "Submit" button. More optional parameters are shown in the following pop-up form, then the user can set the parameters instead of accepting corresponding default values to reduce the computational cost or achieve more accurate result. The detailed information is as follows:- Fitness Cutoff: A threshold for discarding the matching results. The default value '-1.0' means that all the top ranked hits will be reserved in the result list.
- Max Hits: Maximum number of top ranked hits returned.
- GA Match: Use genetic algorithm to optimize the pharmacophore mapped poses.
- Include Exclude Volume: Taking the excluded volume in the pharmacophore model into account.
- Generate Conformers: Multiple conformations of the target molecule are required prior to mapping since adopts semi-rigid pharmacophore mapping protocol. An in-house conformation generation algorithm Cyndi is used as default option.
- Maximum and Minimum Number of Hydrophobic (HB Donor, HB Acceptor, Positive/Negative Charged Center): will skip the pharmacophore model without mapping, if it possesses less features than the user defined.
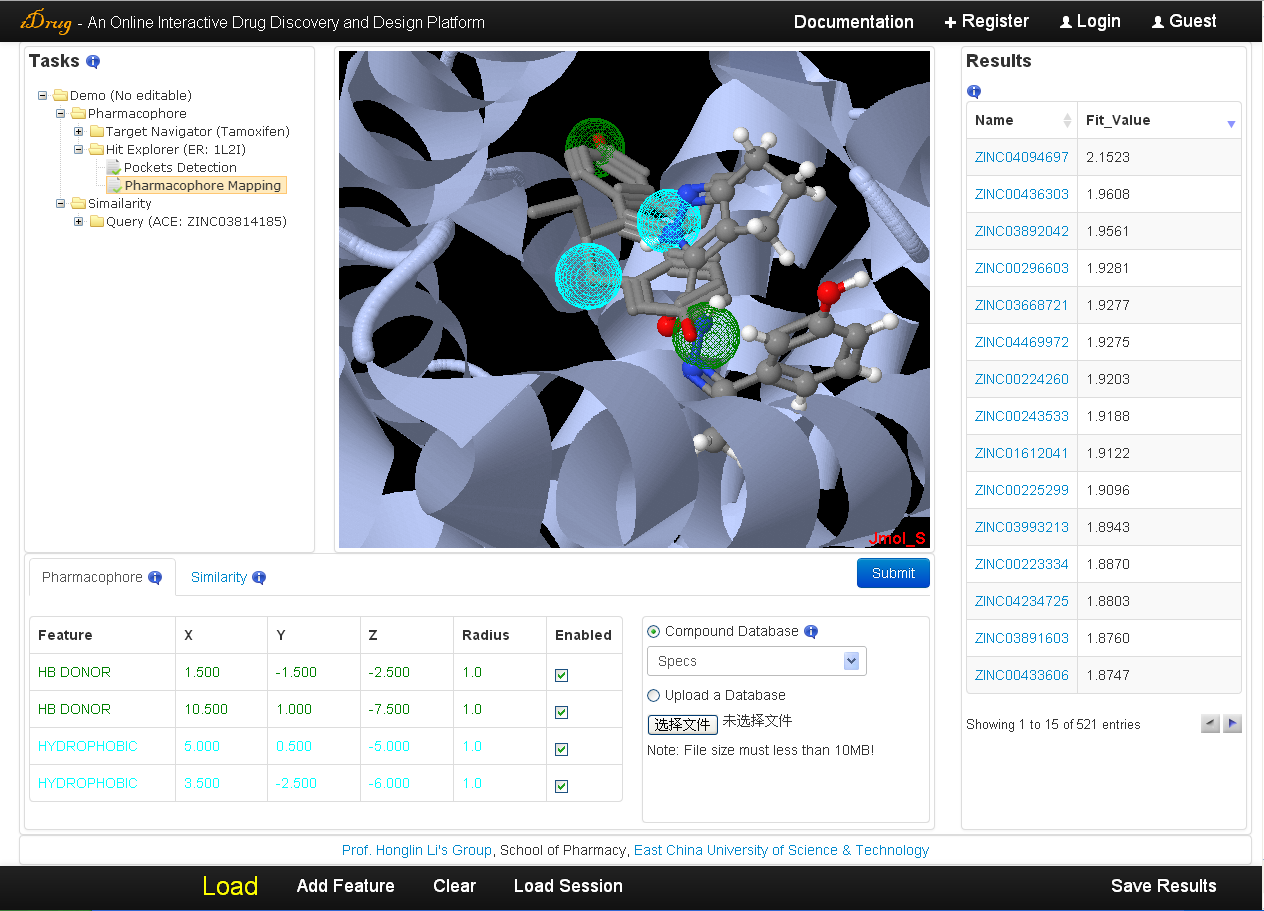
3.4 Results visualization
The user may check the searching results when the job is completed. The search time of iDrug depends on the flexibility of the pharmacophore query, the mapping parameters and target database assigned by the user. A list of hit compounds ranked by fit value is returned and displayed in the result table. The detailed information is as follows:- The unique identifier shown in the result table is a hyperlink to the compound of corresponding chemical database for extra information. Moreover, each hit can be visualized in Jmol by clicking the relevant row in the "Fit Value" column.
- In the Jmol applet panel, all query information is shown by default, including the user-uploaded molecular files and the pharmacophore query shown as spheres.
- In the pharmacophore query editor panel, the query features are listed in the table, which can be edited and resubmitted for more accurate screening.
- The user can download the complete set of hits in a mol2 file through the "Save Results" button for off-line analysis.
NOTE: iDrug will take 10-20 hours to perform pharmacophore matching against the available database!
-
4. Target Identification
- iDrug uses the reverse pharmacophore mapping approach to flexibly align the given molecule onto pharmacophore models of protein in the target list. An example of target identification of the marketed selective estrogen receptor inhibitor Tamoxifen is displayed in the Demo.
-
4.1 Query molecule preparation
The input file consists of only the test molecular structure in standard mol2 format. The user may click on the "Load" button and select the "Pharmacophore" work type. In the "Target Navigator" frame, upload a desired compound and press the "Submit" button, then a job calculating the pharmacophore model of the query molecule will be created.
4.2 Searching database preparation
iDrug offers a large, in-house repertoire of pharmacophore database (PharmTargetDB), which contains over 7,000 pharmacophore models (2241 entries are annotated as "Human protein targets") with target information extracted from DrugBank, PDBSum, UniProt and in-house TargetBank. The target protein structures co-complexed with small molecules to extract pharmacophore models are selected from DrugBank, BindingDB, PDBBind and our PDTD databases.
The user can check the pharmacophore features of the uploaded structure when the job is completed. The pharmacophore query editor in the bottom left will be changed when "Target Navigator" or the corresponding job is selected. In the "Select Targets Set" frame, "Human Targets Only" is available if the user wants to focus on the human protein targets.
4.3 Mapping parameters customization
After selecting the target pharmacophore database, press the "Submit" button to set more optional parameters like:- Generate Conformers: an in-house program Cyndi is used by default to generate multiple conformations or the users can skip this by uploading pre-generated conformations.
- Maximum Generated Conformations: maximum conformations generated by Cyndi.
- Number of Reserved Matched Targets: maximum number of top ranked target candidates.
- Fit Value Cutoff: discard the matching results if the fit value is below the threshold.
- Minimum number of Hydrophobic Center (HB Donor, HB Acceptor, Positive/Negative Ionizable): will skip the pharmacophore model containing less features than the users defined minimum numbers.
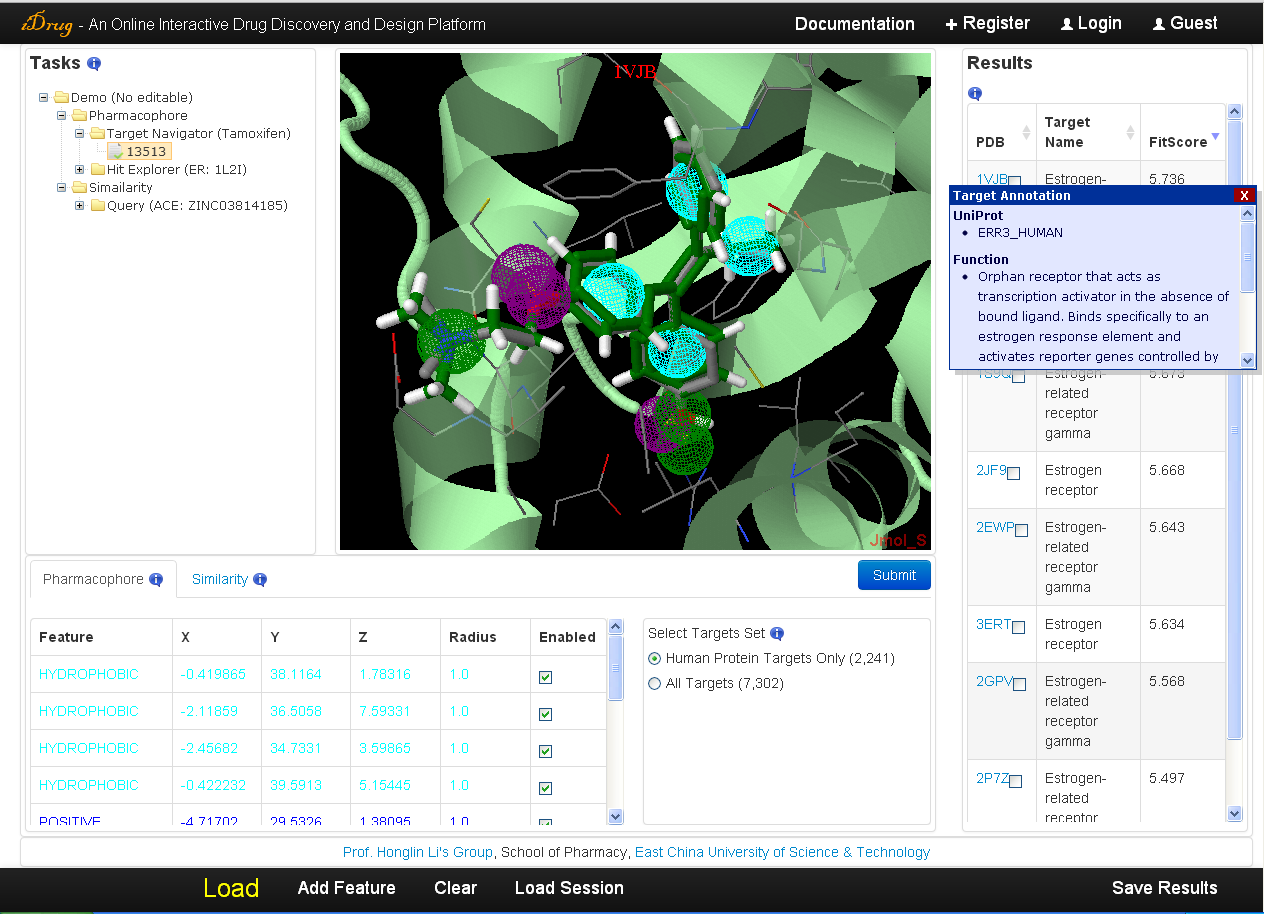
4.4 Results visualization
The user may check the searching results when the job is completed. The output is demonstrated in the result table ranked by fit score. The detailed information is as follows:- In the result table, each target is represented with the PDB ID, target name, and fit score. A hotlink to the PDB website is provided in the "PDB" column and detailed annotations containing the Uniprot identifier, functional and therapeutic information will be displayed in the pop-up window when the cursor moving over the PDB ID.
- All information, including the target structure, the mapping pharmacophore model, the aligned pose of the query molecule, and the residues within 6A of the bound-ligand in the co-complexed protein will be shown in Jmol applet by clicking on the relevant row in the result table. But this may take seconds for Jmol to load receptor files directly from the PDB server.
- The corresponding mapping pharmacophore features are also listed in the pharmacophore query editor.
- The pharmacophore and aligned molecule can be saved by selecting the checkbox in the result table and clicking on the "Save Results" button. The complete targets information in comma separated values (CSV) format will always be included in the downloaded file. Besides, iDrug will spontaneous download all the matched results if none of the targets is selected.
NOTE: iDrug will take 1-2 hours to perform target identification based on reverse pharmacophore mapping approach!
-
5. Similarity Calculation
- iDrug adopts the 3D similarity method SHAFTS to perform ligand-based virtual screening, a hybrid similarity metric combined with chemical feature matching and volumetric overlay. An example of 3D similarity calculation of the EGFR inhibitor HYZ_2RGP, with the searching database collected from DUD-E data sets is displayed in the Demo.
-
5.1 Query molecule preparation
iDrug offers two possible ways to submit a query chemical structure by pressing the "Load" button and selecting the "Similarity" work type:- Upload a small molecule file in mol2 format in the first text box.
- Click "here" to sketch in JME applet or type as a SMILES string in the text box of the pop-up window. The user must convert the 2D molecule drawn in JME into SMILES by clicking on the "Convert to Smiles" button, which Jmol can turn into 3D structure by clicking on the "Load into Jmol" button.
5.2 Searching database selection
In the similarity query editor displayed in the bottom left, the user is expected to select the searching chemical library or upload an in-house small database as the target. The detailed information is described in Section 3.2.
5.3 Filter parameters setting
In the similarity query editor, five thresholds (0.8, 1.0, 1.2, 1.5 and 1.8, with 1.2 as the default value) are supported and all the compounds below the thresholds will be discarded in the searching process.
The maximum number of hit compounds is set as 1000 by default to limit the output size.
After selecting the target database and setting the filter parameters, the user may just press "Submit" to deliver the query molecule and all the parameters to the server to perform similarity calculation.
5.4 Results visualization
A list of compounds sorted by hybrid score in decreasing order is returned by iDrug. The detailed information is as follows:- The user can also re-rank the result list by feature score or shape score via clicking on the corresponding row in the result table. A hyperlink pointing to the compound of the user-selected commercial chemical library is accessible by clicking the molecular identifier.
- Each hit can be visualized with the query molecule, as well as the pharmacophore model and the surface of the query by clicking on the corresponding row in the result table.
- The pharmacophore features are also listed in the pharmacophore query editor, which can be shown/hidden/modified by the user.
- All the matching compounds is available for downloading a compressed file through the "Save Results" button.
NOTE: iDrug will take 1-2 days to perform 3D similarity calculation against the available database!
-
Reference
- Xia Wang, Haipeng Chen, Feng Yang, Jiayu Gong, Shiliang Li, Jianfeng Pei, Xiaofeng Liu, Hualiang Jiang, Luhua Lai, Honglin Li. iDrug: a web-accessible and interactive drug discovery and design platform. J. Cheminform. 2014, 6, 28.
- Xiaofeng Liu, Hualiang Jiang, Honglin Li. SHAFTS: A Hybrid Approach for 3D Molecular Similarity Calculation. 1. Method and Assessment of Virtual Screening. J. Chem. Inf. Model. 2011, 51, 2372-2385.
- Weiqiang Lu, Xiaofeng Liu, Xianwen Cao, Mengzhu Xue, Kangdong Liu, Zhenjiang Zhao, Xu Shen, Hualiang Jiang, Yufang Xu, Jin Huang, and Honglin Li. SHAFTS: A Hybrid Approach for 3D Molecular Similarity Calculation. 2. Prospective Case Study in the Discovery of Diverse p90 Ribosomal S6 Protein Kinase 2 Inhibitors to Suppress Cell Migration. J. Med. Chem. 2011, 54, 3564-3574.
- Xiaofeng Liu, Hua Xie, Cheng Luo, Linjiang Tong, Yi Wang, Ting Peng, Jian Ding, Hualiang Jiang,Honglin Li. Discovery and SAR of Thiazolidine-2,4-dione Analogues as Insulin-like Growth Factor-1 Receptor (IGF-1R) Inhibitors via Hierarchical Virtual Screening. J. Med. Chem. 2010, 53, 2661-2665.
- Jing Chen, Luhua Lai. Pocket v.2: Further Developments on Receptor-Based Pharmacophore Modeling. J. Chem. Inf. Model. 2006, 46, 2684-2691.
- Yaxia Yuan, Jianfeng Pei, Luhua Lai. Binding Site Detection and Druggability Prediction of Protein Targets for Structure-Based Drug Design. Curr. Pharm. Des. 2013, 19, 2326-2333.
- Yaxia Yuan, Jianfeng Pei, Luhua Lai. LigBuilder 2: A Practical de Novo Drug Design Approach. J. Chem. Inf. Model., 2011, 51, 1083-1091.
Prof. Honglin Li's Group
, School of Pharmacy,
East China University of Science & Technology